
Synergistic Cytotoxicity of Histone Deacetylase and Poly-ADP Ribose Polymerase Inhibitors and Decitabine in Breast and Ovarian Cancer Cells: Implications for Novel Therapeutic Combinations

 , , , and
, , , and
Abstract
1. Introduction
2. Results
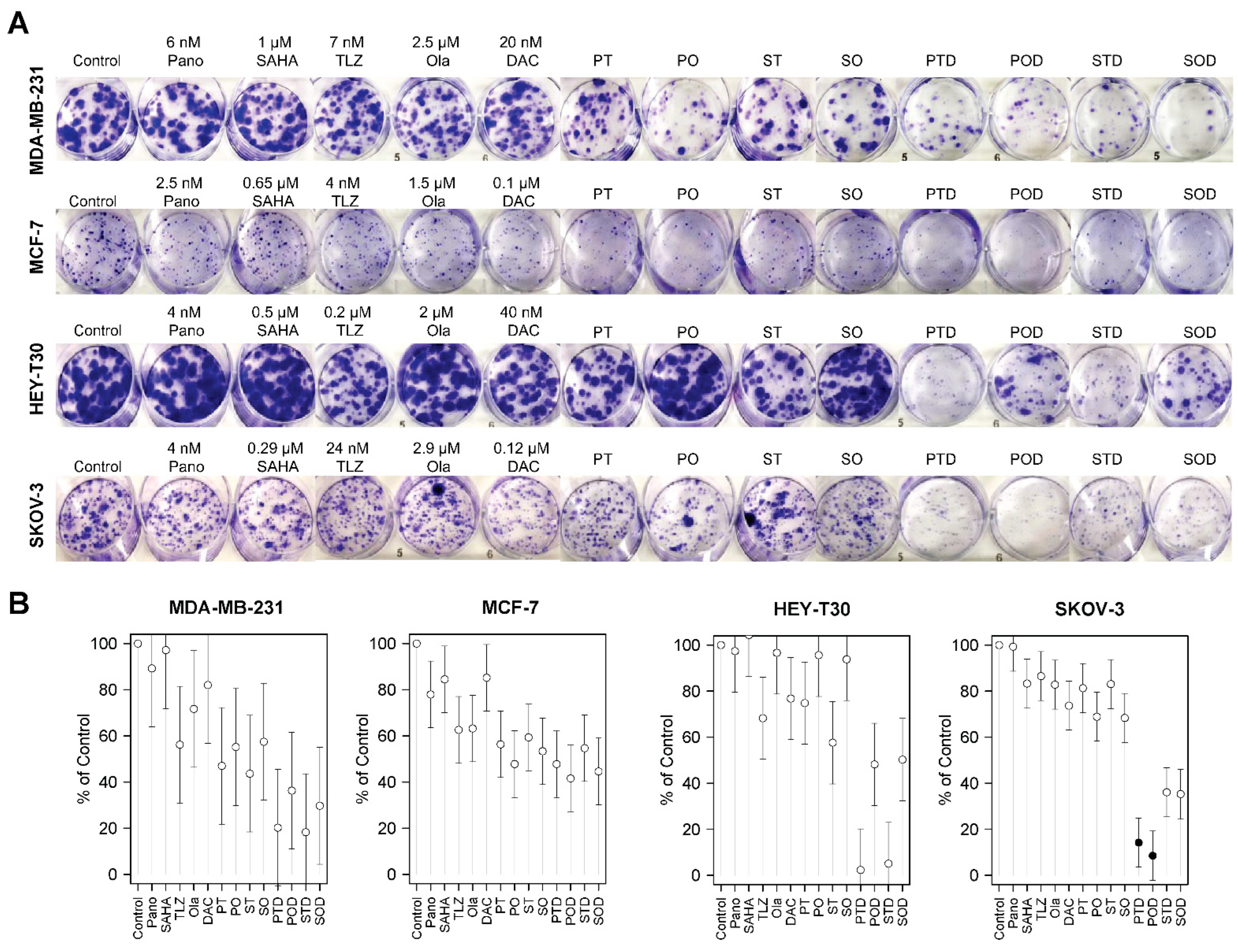
2.1. Sensitivity of Breast and Ovarian Cancer Cell Lines to HDACis, PARPis, and Decitabine
2.2. Synergistic Cytotoxicity of HDACis, PARPis, and Decitabine in Breast and Ovarian Cancer Cell Lines
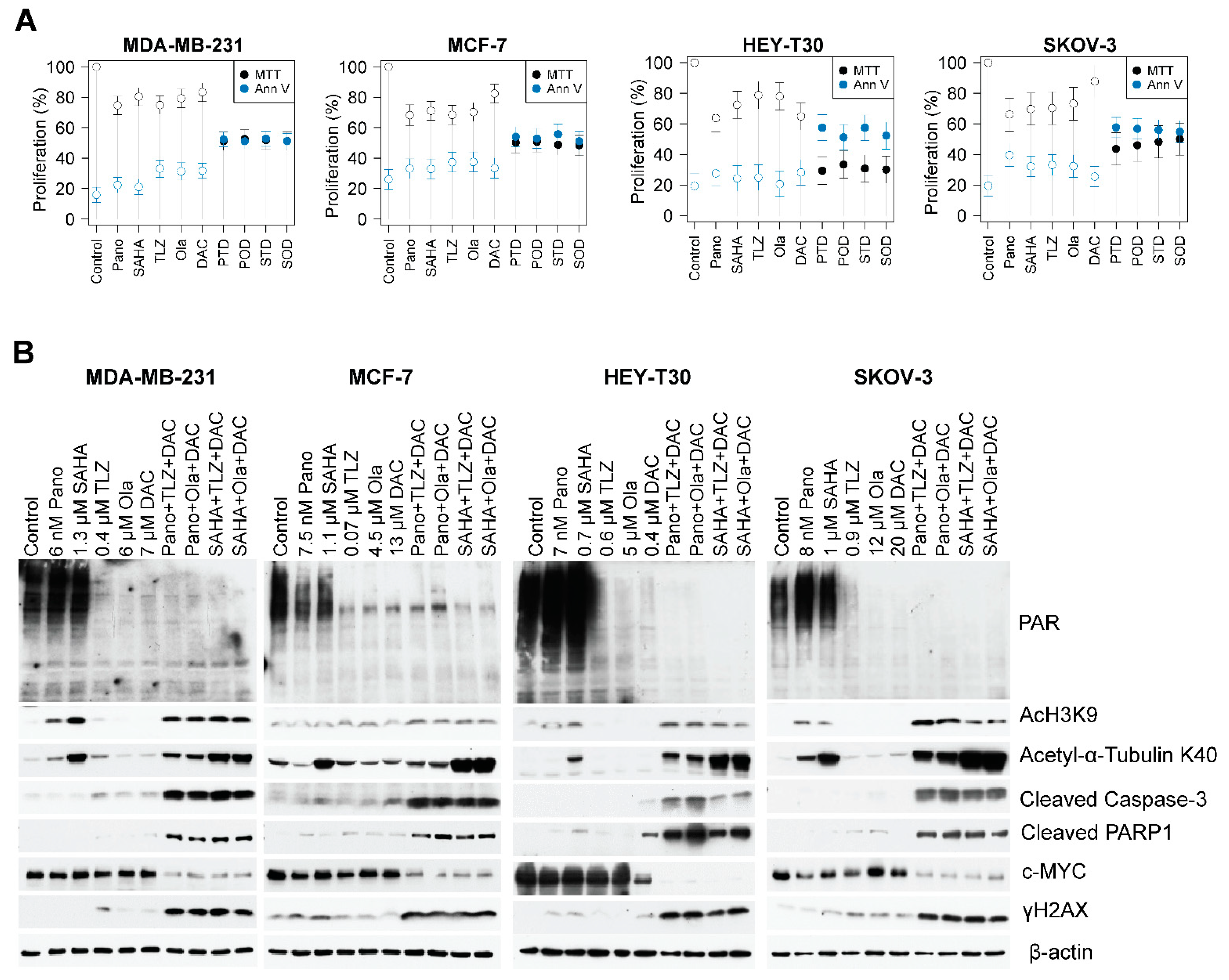
2.3. Drug-Mediated Inhibition of Cell Proliferation and PARylation, and Effects on Survival and Apoptosis Protein Markers
2.4. The Three-Drug Combinations Inhibit PARylation and Enhance Cleavage of Caspase 3 and PARP1
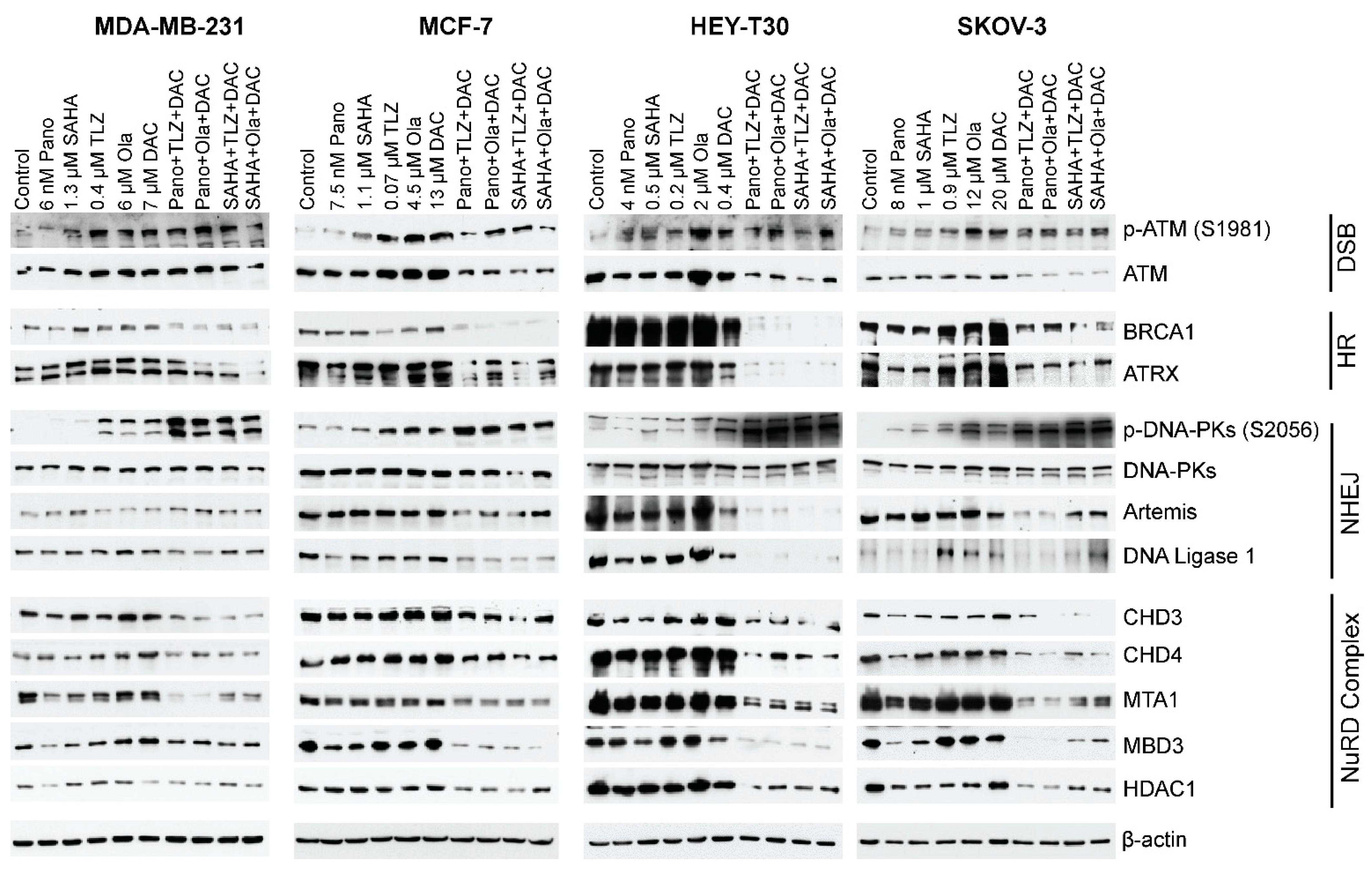
2.5. HDACi, PARPi, and Decitabine Combinations Affect the Levels of Proteins Involved in DNA Damage Response and Repair
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Drugs
4.2. Determination of IC50 and Drug Synergism
4.3. Colony Formation/Clonogenic Assay
4.4. MTT Assay and Western Blot Analysis
4.5. Drug Concentrations for Colony Formation/Clonogenic Assay, MTT Assay, and Western Blot Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATM | Ataxia–Telangiectasia Mutated protein |
| ATRX | Alpha Thalassemia/Mental Retardation Syndrome X-Linked protein |
| BRCA | Breast Cancer gene |
| c-MYC | Cellular Myelocytomatosis oncogene |
| CI | Combination Index (used in drug synergism analysis) |
| DCIS | Ductal Carcinoma In Situ |
| DNAPKcs | DNA-Dependent Protein Kinase Catalytic Subunit |
| Fa | Fractions affected (refers to cell death in drug synergism analysis) |
| FDA | U.S. Food and Drug Administration |
| HDACis | Histone Deacetylase Inhibitors |
| IC50 | Half-Maximal Inhibitory Concentration |
| JAK–STAT | Janus Kinase–Signal Transducer and Activator of Transcription |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyl Tetrazolium Bromide |
| NuRD | Nucleosome Remodeling and Deacetylase complex |
| PARPis | Poly(ADP Ribose) Polymerase Inhibitors |
| PD-L1 | Programmed Death (Ligand) 1 |
| PBS | Phosphate-Buffered Saline |
| TNF | Tumor Necrosis Factor |
| UHFR1 | Ubiquitin-Specific |
| Histone | H2A and H2B Deubiquitinase Factor R1 |
| γ-H2AX | Phosphorylated Histone H2AX |
References
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Key Statistics for Breast Cancer. Available online: https://www.cancer.org/cancer/types/breast-cancer/about/how-common-is-breast-cancer.html (accessed on 29 January 2024).
- American Cancer Society. Key Statistics for Breast Cancer in Men. Available online: https://www.cancer.org/cancer/types/breast-cancer-in-men/about/key-statistics.html (accessed on 29 January 2024).
- American Cancer Society. Key Statistics for Ovarian Cancer. Available online: https://www.cancer.org/cancer/types/ovarian-cancer/about/key-statistics.html (accessed on 29 January 2024).
- Matei, D.E.; Nephew, K.P. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Kozono, D.; Yang, X.; Fendler, W.; Fitts, W.; Ni, J.; Alberta, J.A.; Zhao, J.; Liu, K.X.; Bian, J.; et al. Dual HDAC and PI3K Inhibition Abrogates NFkappaB- and FOXM1-Mediated DNA Damage Response to Radiosensitize Pediatric High-Grade Gliomas. Cancer Res. 2018, 78, 4007–4021. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Chao, O.S.; Goodman, O.B., Jr. Synergistic loss of prostate cancer cell viability by coinhibition of HDAC and PARP. Mol. Cancer Res. 2014, 12, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.L.; Tamayo, P.; Wilson, A.J.; Wang, S.; Chang, Y.M.; Kim, J.W.; Khabele, D.; Shamji, A.F.; Schreiber, S.L. KRAS Genomic Status Predicts the Sensitivity of Ovarian Cancer Cells to Decitabine. Cancer Res. 2015, 75, 2897–2906. [Google Scholar] [CrossRef]
- Dahn, M.L.; Cruickshank, B.M.; Jackson, A.J.; Dean, C.; Holloway, R.W.; Hall, S.R.; Coyle, K.M.; Maillet, H.; Waisman, D.M.; Goralski, K.B.; et al. Decitabine Response in Breast Cancer Requires Efficient Drug Processing and Is Not Limited by Multidrug Resistance. Mol. Cancer Ther. 2020, 19, 1110–1122. [Google Scholar] [CrossRef]
- Raynal, N.J.; Si, J.; Taby, R.F.; Gharibyan, V.; Ahmed, S.; Jelinek, J.; Estecio, M.R.; Issa, J.P. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res. 2012, 72, 1170–1181. [Google Scholar] [CrossRef]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef]
- Al-Romaih, K.; Somers, G.R.; Bayani, J.; Hughes, S.; Prasad, M.; Cutz, J.C.; Xue, H.; Zielenska, M.; Wang, Y.; Squire, J.A. Modulation by decitabine of gene expression and growth of osteosarcoma U2OS cells in vitro and in xenografts: Identification of apoptotic genes as targets for demethylation. Cancer Cell Int. 2007, 7, 14. [Google Scholar] [CrossRef]
- Cooper, S.J.; von Roemeling, C.A.; Kang, K.H.; Marlow, L.A.; Grebe, S.K.; Menefee, M.E.; Tun, H.W.; Colon-Otero, G.; Perez, E.A.; Copland, J.A. Reexpression of tumor suppressor, sFRP1, leads to antitumor synergy of combined HDAC and methyltransferase inhibitors in chemoresistant cancers. Mol. Cancer Ther. 2012, 11, 2105–2115. [Google Scholar] [CrossRef]
- Lubbert, M.; Suciu, S.; Baila, L.; Ruter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R.; et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: Final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J. Clin. Oncol. 2011, 29, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Huang, F.X.; Wei, W.; Li, Q.Q.; Wu, J.W.; Huang, Y.; Li, Z.L.; Zhang, H.L.; Li, X.; Yuan, Q.E.; et al. Loss of HRD functional phenotype impedes immunotherapy and can be reversed by HDAC inhibitor in ovarian cancer. Int. J. Biol. Sci. 2023, 19, 1846–1860. [Google Scholar] [CrossRef]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef]
- Meng, F.; Sun, G.; Zhong, M.; Yu, Y.; Brewer, M.A. Inhibition of DNA methyltransferases, histone deacetylases and lysine-specific demethylase-1 suppresses the tumorigenicity of the ovarian cancer ascites cell line SKOV3. Int. J. Oncol. 2013, 43, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Fang, F.; Shen, C.; Schilder, J.; Arnold, A.; Zeng, Y.; Berry, W.A.; Huang, T.; Nephew, K.P. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012, 72, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637–5650. [Google Scholar] [CrossRef]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L.; et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 153. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Safari, M.; Litman, T.; Robey, R.W.; Aguilera, A.; Chakraborty, A.R.; Reinhold, W.C.; Basseville, A.; Petrukhin, L.; Scotto, L.; O’Connor, O.A.; et al. R-Loop-Mediated ssDNA Breaks Accumulate Following Short-Term Exposure to the HDAC Inhibitor Romidepsin. Mol. Cancer Res. 2021, 19, 1361–1374. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, J. Targeted Cancer Therapy Based on Acetylation and Deacetylation of Key Proteins Involved in Double-Strand Break Repair. Cancer Manag. Res. 2022, 14, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Valdez, B.C.; Nieto, Y.; Yuan, B.; Murray, D.; Andersson, B.S. HDAC inhibitors suppress protein poly(ADP-ribosyl)ation and DNA repair protein levels and phosphorylation status in hematologic cancer cells: Implications for their use in combination with PARP inhibitors and chemotherapeutic drugs. Oncotarget 2022, 13, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Protein ADP-ribosylation and the cellular response to DNA strand breaks. DNA Repair 2014, 19, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Tallis, M.; Morra, R.; Barkauskaite, E.; Ahel, I. Poly(ADP-ribosyl)ation in regulation of chromatin structure and the DNA damage response. Chromosoma 2014, 123, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Gursoy-Yuzugullu, O.; House, N.; Price, B.D. Patching Broken DNA: Nucleosome Dynamics and the Repair of DNA Breaks. J. Mol. Biol. 2016, 428, 1846–1860. [Google Scholar] [CrossRef]
- Smeenk, G.; Wiegant, W.W.; Vrolijk, H.; Solari, A.P.; Pastink, A.; van Attikum, H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol. 2010, 190, 741–749. [Google Scholar] [CrossRef]
- Hou, M.F.; Luo, C.W.; Chang, T.M.; Hung, W.C.; Chen, T.Y.; Tsai, Y.L.; Chai, C.Y.; Pan, M.R. The NuRD complex-mediated p21 suppression facilitates chemoresistance in BRCA-proficient breast cancer. Exp. Cell Res. 2017, 359, 458–465. [Google Scholar] [CrossRef]
- Chan, D.W.; Lees-Miller, S.P. The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J. Biol. Chem. 1996, 271, 8936–8941. [Google Scholar] [CrossRef]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef]
- Liszczak, G.; Diehl, K.L.; Dann, G.P.; Muir, T.W. Acetylation blocks DNA damage-induced chromatin ADP-ribosylation. Nat. Chem. Biol. 2018, 14, 837–840. [Google Scholar] [CrossRef]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Cole, P.A.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk. Res. 2016, 45, 14–23. [Google Scholar] [CrossRef]
- Chen, M.K. Efficacy of PARP inhibition combined with EZH2 inhibition depends on BRCA mutation status and microenvironment in breast cancer. FEBS J. 2021, 288, 2884–2887. [Google Scholar] [CrossRef] [PubMed]
- Masuda, N.; Bando, H.; Yamanaka, T.; Kadoya, T.; Takahashi, M.; Nagai, S.E.; Ohtani, S.; Aruga, T.; Suzuki, E.; Kikawa, Y.; et al. Eribulin-based neoadjuvant chemotherapy for triple-negative breast cancer patients stratified by homologous recombination deficiency status: A multicenter randomized phase II clinical trial. Breast Cancer Res. Treat. 2021, 188, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Dai, Q.; Zhao, L.; Zheng, C.; Li, Q.; Yuan, Z.; Li, L.; Xie, Z.; Qiu, Z.; Huang, W.; et al. Novel dual inhibitors of PARP and HDAC induce intratumoral STING-mediated antitumor immunity in triple-negative breast cancer. Cell Death Dis. 2024, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Mani, C.; Reddy, P.H.; Palle, K. DNA repair fidelity in stem cell maintenance, health, and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165444. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Wilson, A.J.; Saskowski, J.; Wass, E.; Khabele, D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol. Oncol. 2014, 133, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Soria, J.C.; Lord, C.J.; Postel-Vinay, S. Beyond DNA repair: The novel immunological potential of PARP inhibitors. Mol. Cell Oncol. 2019, 6, 1585170. [Google Scholar] [CrossRef]
- Gatla, H.R.; Zou, Y.; Uddin, M.M.; Singha, B.; Bu, P.; Vancura, A.; Vancurova, I. Histone Deacetylase (HDAC) Inhibition Induces IkappaB Kinase (IKK)-dependent Interleukin-8/CXCL8 Expression in Ovarian Cancer Cells. J. Biol. Chem. 2017, 292, 5043–5054. [Google Scholar] [CrossRef]
- Gross, S.M.; Mohammadi, F.; Sanchez-Aguila, C.; Zhan, P.J.; Liby, T.A.; Dane, M.A.; Meyer, A.S.; Heiser, L.M. Analysis and modeling of cancer drug responses using cell cycle phase-specific rate effects. Nat. Commun. 2023, 14, 3450. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The Receptor Tyrosine Kinase AXL in Cancer Progression. Cancers 2016, 8, 103. [Google Scholar] [CrossRef]
- Colavito, S.A. AXL as a Target in Breast Cancer Therapy. J. Oncol. 2020, 2020, 5291952. [Google Scholar] [CrossRef] [PubMed]
- Balaji, K.; Vijayaraghavan, S.; Diao, L.; Tong, P.; Fan, Y.; Carey, J.P.; Bui, T.N.; Warner, S.; Heymach, J.V.; Hunt, K.K.; et al. AXL Inhibition Suppresses the DNA Damage Response and Sensitizes Cells to PARP Inhibition in Multiple Cancers. Mol. Cancer Res. 2017, 15, 45–58. [Google Scholar] [CrossRef]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Mikubo, M.; Inoue, Y.; Liu, G.; Tsao, M.S. Mechanism of Drug Tolerant Persister Cancer Cells: The Landscape and Clinical Implication for Therapy. J. Thorac. Oncol. 2021, 16, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Woodgate, R. What a difference a decade makes: Insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 15591–15598. [Google Scholar] [CrossRef]
- Vaziri, C.; Rogozin, I.B.; Gu, Q.; Wu, D.; Day, T.A. Unravelling roles of error-prone DNA polymerases in shaping cancer genomes. Oncogene 2021, 40, 6549–6565. [Google Scholar] [CrossRef]
- Ziv, O.; Zeisel, A.; Mirlas-Neisberg, N.; Swain, U.; Nevo, R.; Ben-Chetrit, N.; Martelli, M.P.; Rossi, R.; Schiesser, S.; Canman, C.E.; et al. Identification of novel DNA-damage tolerance genes reveals regulation of translesion DNA synthesis by nucleophosmin. Nat. Commun. 2014, 5, 5437. [Google Scholar] [CrossRef]
- Welsh, J. Chapter 40—Animal Models for Studying Prevention and Treatment of Breast Cancer. In Animal Models for the Study of Human Disease; Conn, P.M., Ed.; Academic Press: Boston, MA, USA, 2013; pp. 997–1018. [Google Scholar]
- Comsa, S.; Cimpean, A.M.; Raica, M. The Story of MCF-7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer Res. 2015, 35, 3147–3154. [Google Scholar]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Lenth, R. Estimated Marginal Means, aka Least-Squares Means. Available online: https://CRAN.R-project.org/package=emmeans (accessed on 20 January 2024).
- José, C.P.; Douglas, M.B. Mixed-Effects Models in S and S-PLUS. Available online: https://link.springer.com/book/10.1007/b98882 (accessed on 20 January 2024).
- José, P.; Douglas, B.; R Core Team. Linear and Nonlinear Mixed Effects Models. Available online: https://CRAN.R-project.org/package=nlme (accessed on 20 January 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breast Cancer Cell Lines | ||||||||||
| MDA-MB-231 | MCF-7 | |||||||||
| Pano | SAHA | TLZ | OLA | DAC | Pano | SAHA | TLZ | OLA | DAC | |
| Cell Proliferation | ||||||||||
| PTD | <0.0001 | <0.0001 | <0.0001 | 0.003 | 0.003 | <0.0001 | ||||
| POD | <0.0001 | <0.0001 | <0.0001 | 0.003 | 0.0009 | <0.0001 | ||||
| STD | <0.0001 | <0.0001 | <0.0001 | 0.0001 | 0.001 | <0.0001 | ||||
| SOD | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.0002 | <0.0001 | ||||
| Apoptosis | ||||||||||
| PTD | <0.0001 | 0.0001 | <0.0001 | 0.011 | 0.068 | 0.014 | ||||
| POD | <0.0001 | 0.0001 | 0.0001 | 0.021 | 0.1 | 0.022 | ||||
| STD | <0.0001 | 0.0001 | <0.0001 | 0.004 | 0.031 | 0.005 | ||||
| SOD | <0.0001 | 0.0001 | 0.0001 | 0.036 | 0.13 | 0.046 | ||||
| Ovarian Cancer Cell Lines | ||||||||||
| HEY-T30 | SKOV-3 | |||||||||
| Pano | SAHA | TLZ | OLA | DAC | Pano | SAHA | TLZ | OLA | DAC | |
| Cell Proliferation | ||||||||||
| PTD | <0.0001 | <0.0001 | <0.0001 | 0.0004 | <0.0001 | <0.0001 | ||||
| POD | <0.0001 | <0.0001 | <0.0001 | 0.0008 | <0.0001 | <0.0001 | ||||
| STD | <0.0001 | <0.0001 | <0.0001 | 0.0005 | 0.0005 | <0.0001 | ||||
| SOD | <0.0001 | <0.0001 | <0.0001 | 0.0008 | 0.0003 | <0.0001 | ||||
| Apoptosis | ||||||||||
| PTD | 0.0008 | 0.0003 | 0.001 | 0.037 | 0.001 | <0.0001 | ||||
| POD | 0.0101 | 0.0006 | 0.01 | 0.037 | 0.002 | <0.0001 | ||||
| STD | 0.0002 | 0.0003 | 0.001 | 0.002 | 0.003 | <0.0001 | ||||
| SOD | 0.004 | 0.0006 | 0.01 | 0.006 | 0.008 | 0.0001 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdez, B.C.; Tsimberidou, A.M.; Yuan, B.; Baysal, M.A.; Chakraborty, A.; Andersen, C.R.; Andersson, B.S. Synergistic Cytotoxicity of Histone Deacetylase and Poly-ADP Ribose Polymerase Inhibitors and Decitabine in Breast and Ovarian Cancer Cells: Implications for Novel Therapeutic Combinations. Int. J. Mol. Sci. 2024, 25, 9241. https://doi.org/10.3390/ijms25179241
Valdez BC, Tsimberidou AM, Yuan B, Baysal MA, Chakraborty A, Andersen CR, Andersson BS. Synergistic Cytotoxicity of Histone Deacetylase and Poly-ADP Ribose Polymerase Inhibitors and Decitabine in Breast and Ovarian Cancer Cells: Implications for Novel Therapeutic Combinations. International Journal of Molecular Sciences. 2024; 25(17):9241. https://doi.org/10.3390/ijms25179241
Chicago/Turabian StyleValdez, Benigno C., Apostolia M. Tsimberidou, Bin Yuan, Mehmet A. Baysal, Abhijit Chakraborty, Clark R. Andersen, and Borje S. Andersson. 2024. "Synergistic Cytotoxicity of Histone Deacetylase and Poly-ADP Ribose Polymerase Inhibitors and Decitabine in Breast and Ovarian Cancer Cells: Implications for Novel Therapeutic Combinations" International Journal of Molecular Sciences 25, no. 17: 9241. https://doi.org/10.3390/ijms25179241
APA StyleValdez, B. C., Tsimberidou, A. M., Yuan, B., Baysal, M. A., Chakraborty, A., Andersen, C. R., & Andersson, B. S. (2024). Synergistic Cytotoxicity of Histone Deacetylase and Poly-ADP Ribose Polymerase Inhibitors and Decitabine in Breast and Ovarian Cancer Cells: Implications for Novel Therapeutic Combinations. International Journal of Molecular Sciences, 25(17), 9241. https://doi.org/10.3390/ijms25179241