
Maternal Undernutrition Affects Fetal Thymus DNA Methylation, Gene Expression, and, Thereby, Metabolism and Immunopoiesis in Wagyu (Japanese Black) Cattle

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Effect of MNR on the Thymic Phenotype of the Calves
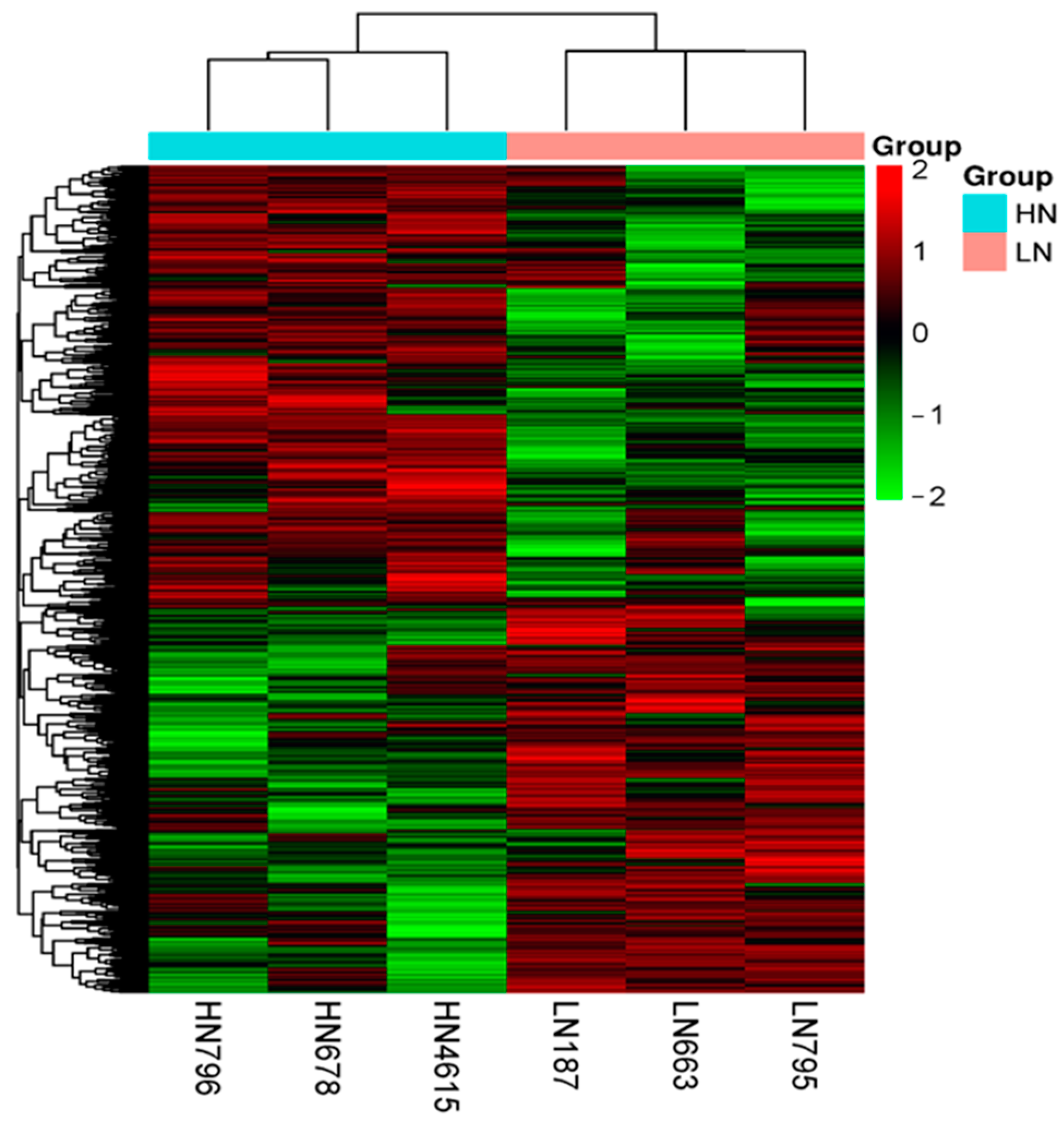
2.2. DNA Methylation Profile
2.2.1. Landscape of DNA Methylation
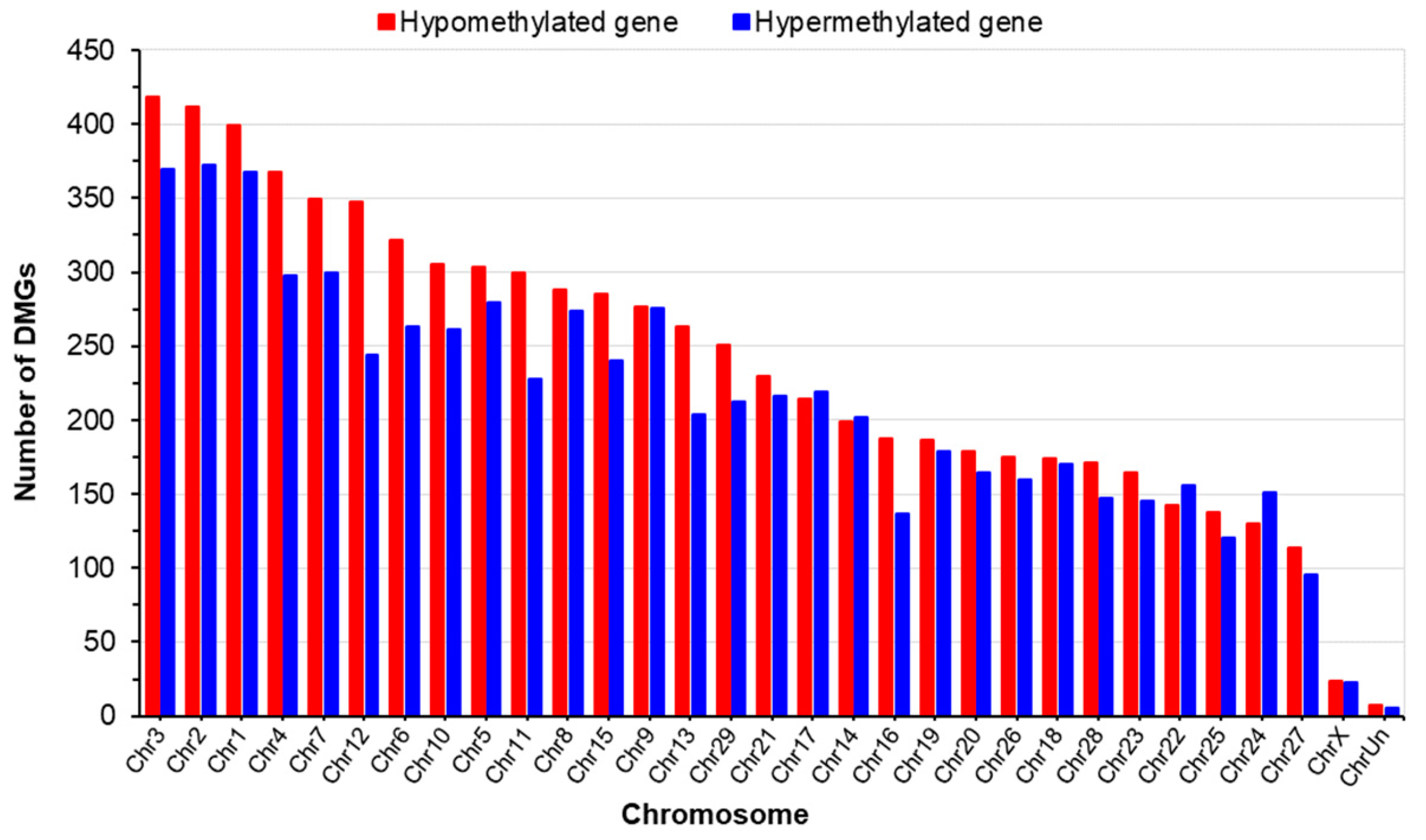
2.2.2. Pattern of DNA Methylation (DMR and DMG)
2.2.3. Functional Enrichment of DMGs
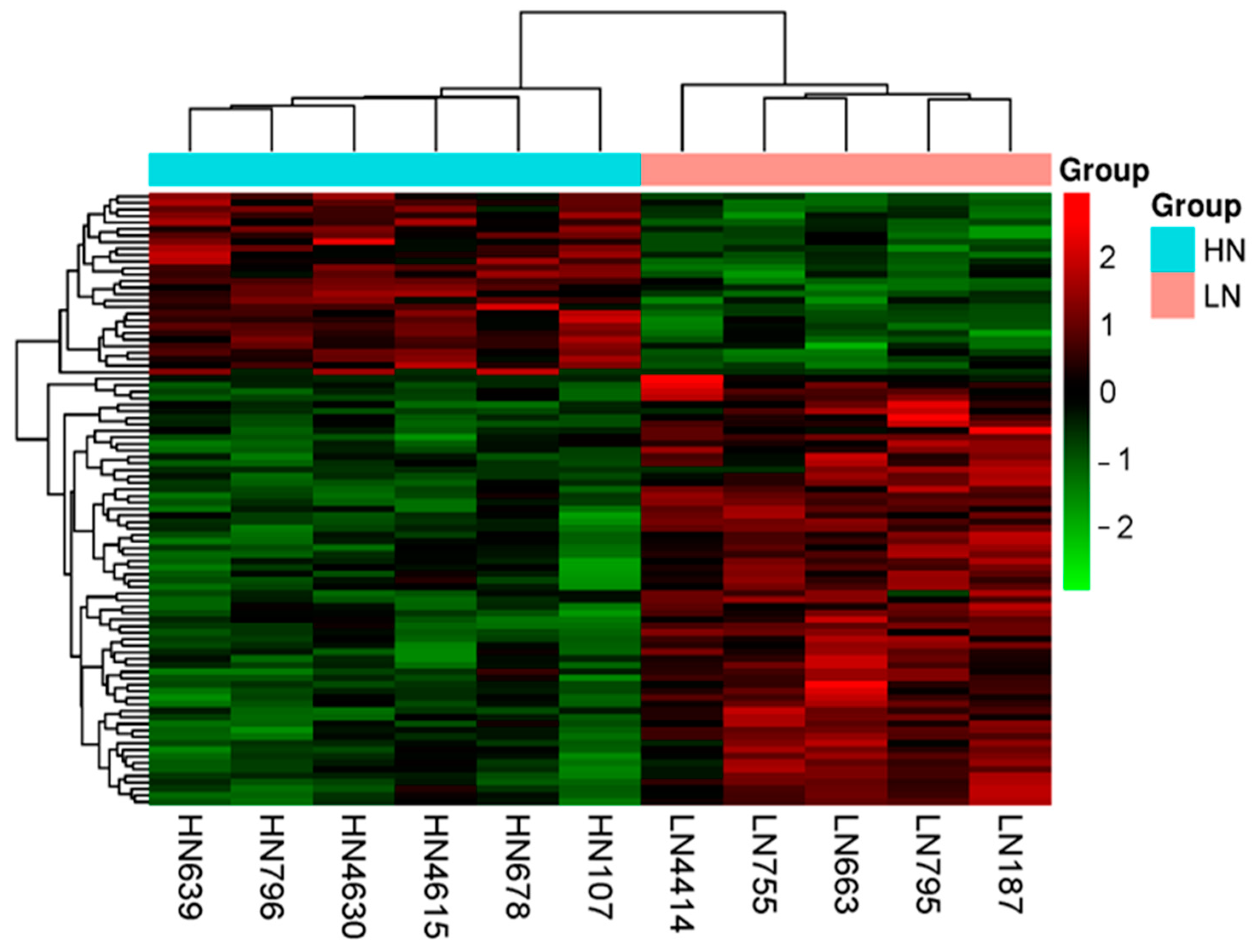
2.3. RNA Sequencing Profiles
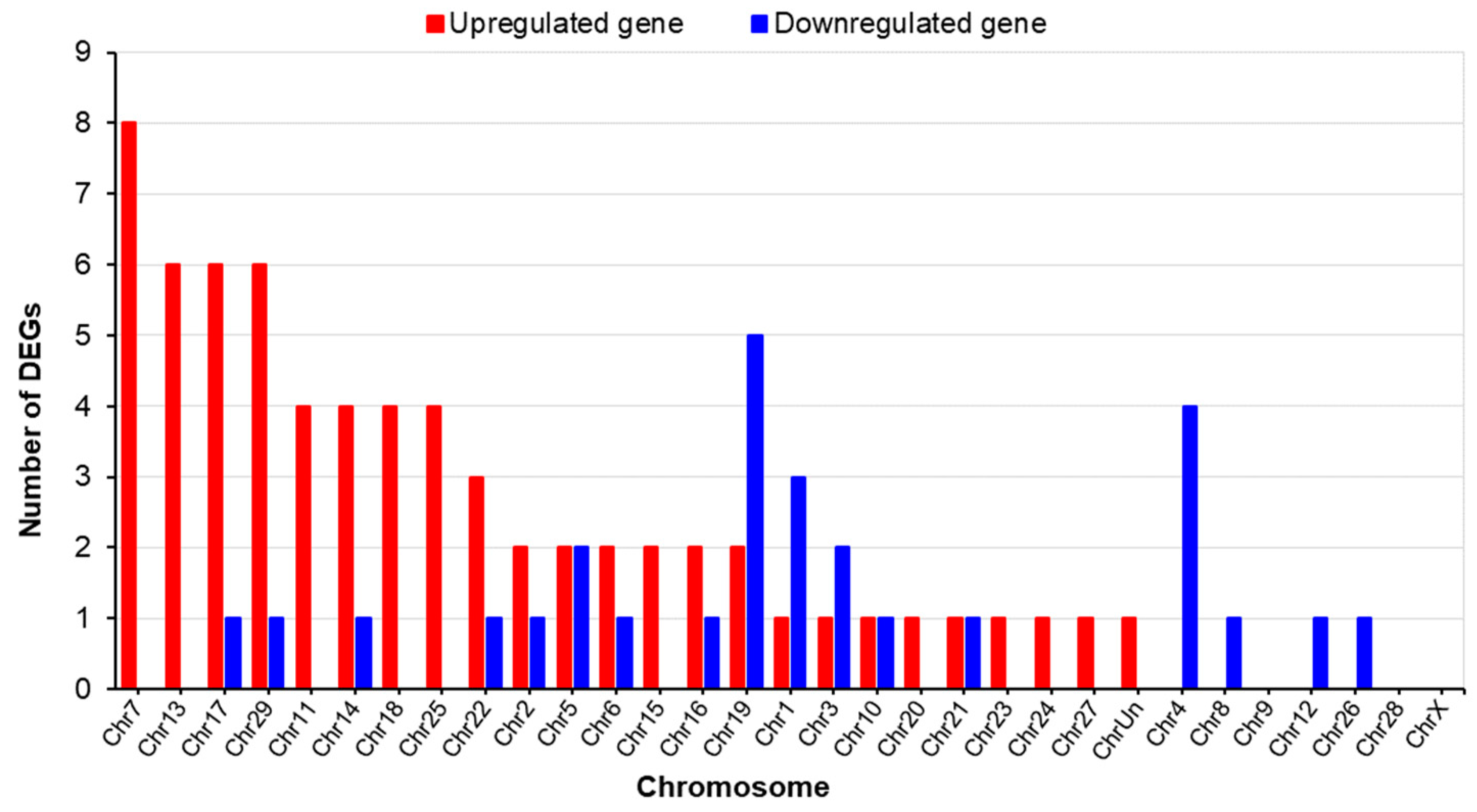
2.3.1. Differences in the Gene Expression of LN and HN Fetal Thymuses
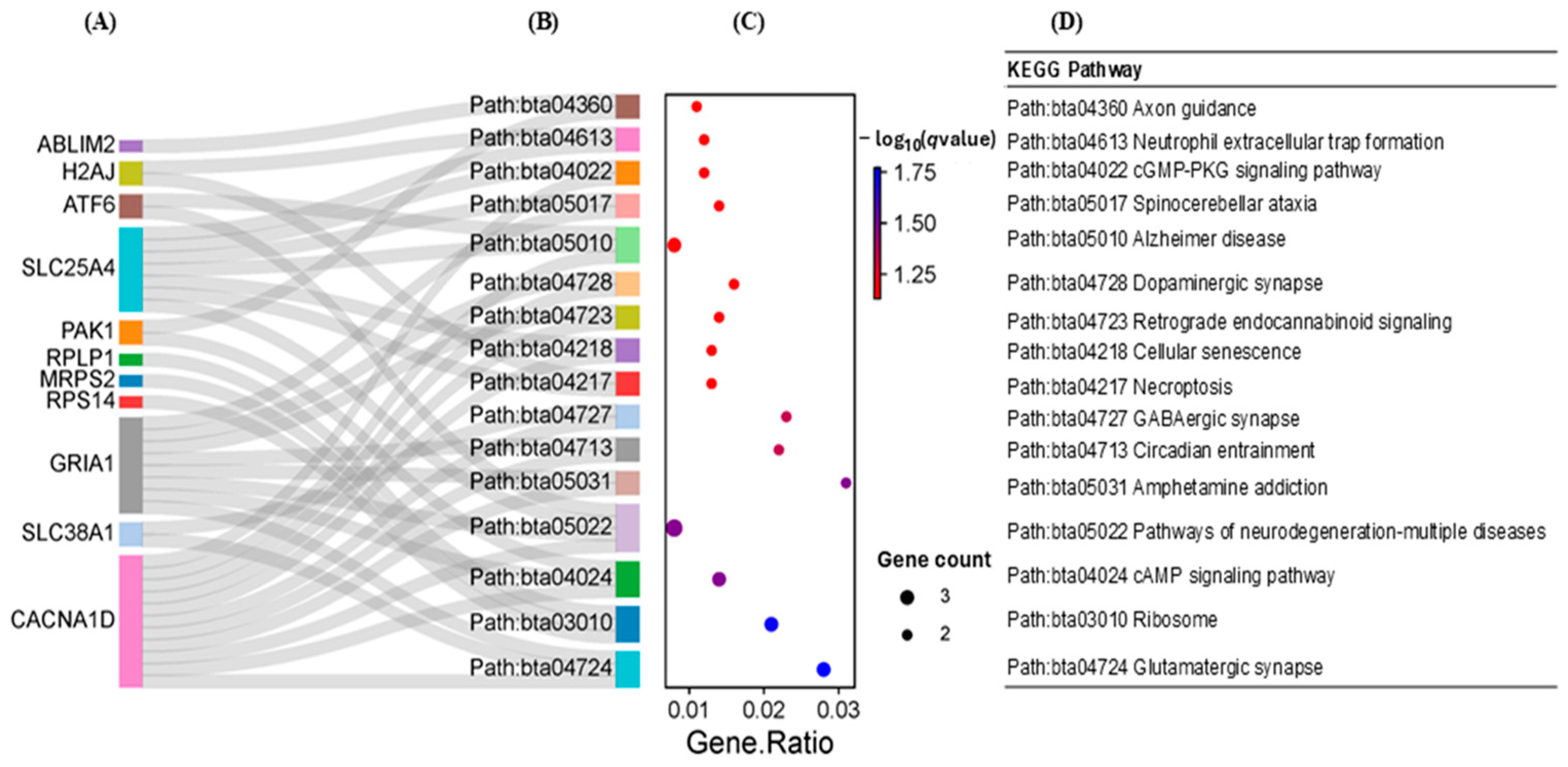
2.3.2. Results of the Functional Enrichment Analysis of the DEGs
2.4. Results of the Overlapping DMG/DEG Gene Analyses of DNA Methylation and Gene Expression
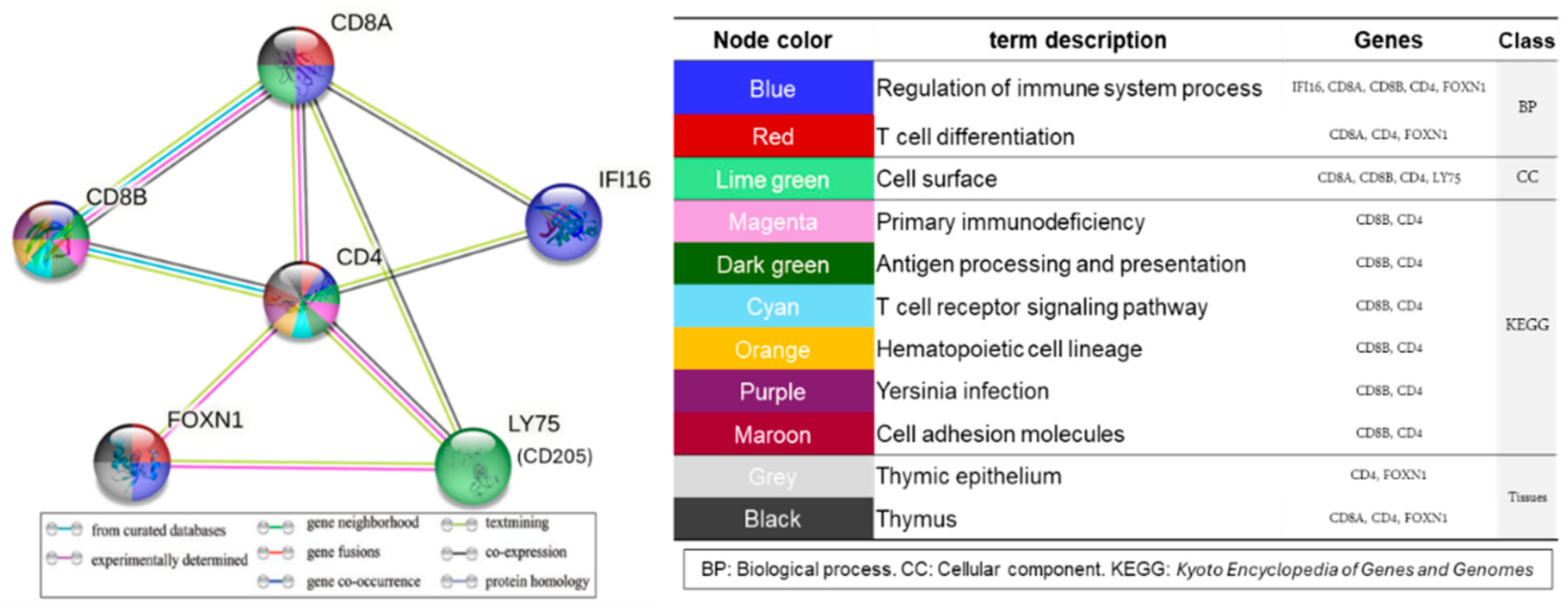
2.5. Immunity-Related Genes Affected by MNR in the Fetal Thymus
Protein–Protein Interaction (PPI) Network Analysis of the Target Genes
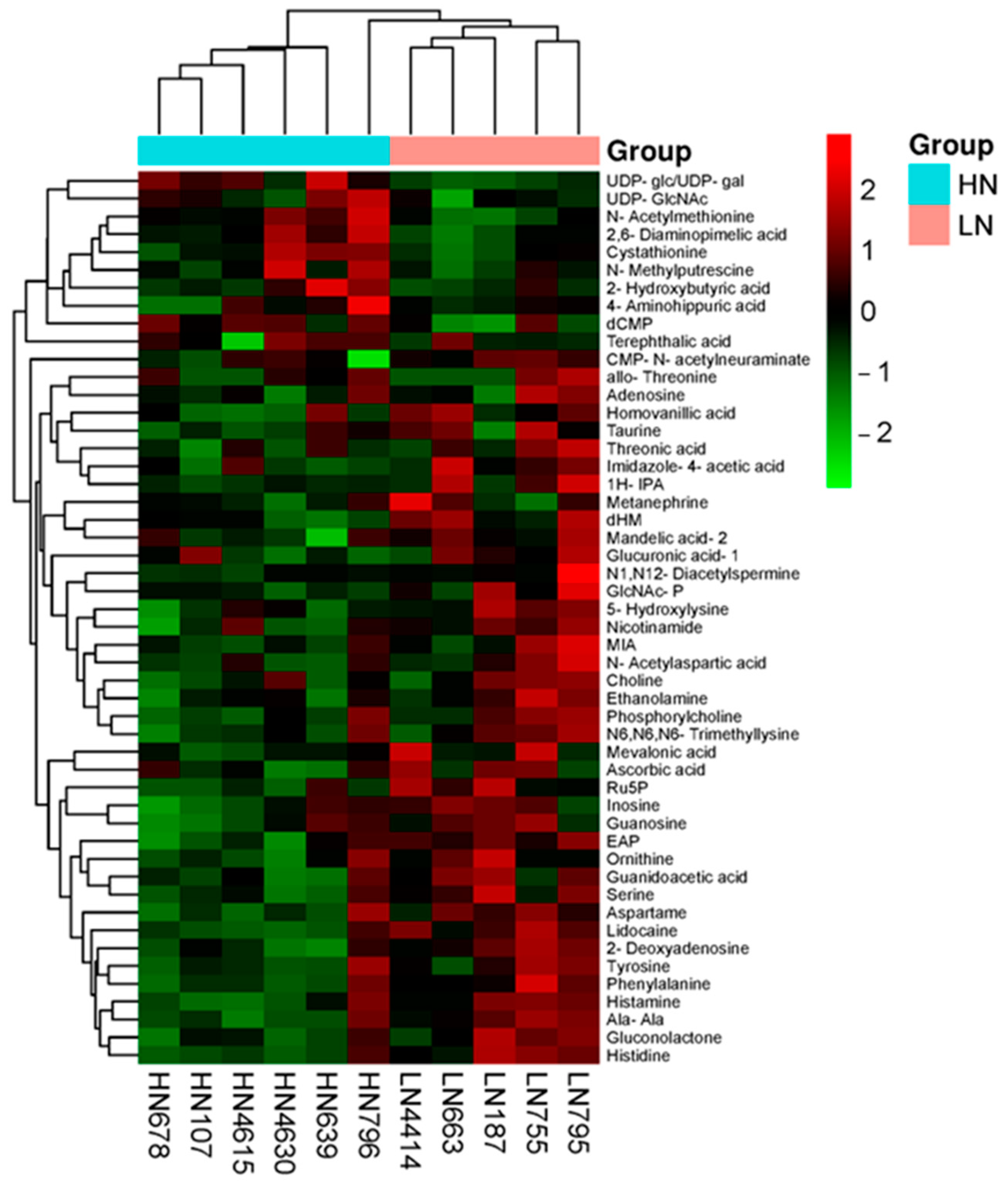
2.6. Metabolomic Profile
3. Discussion
3.1. MNR Affects the Phenotype of the Fetal Thymus
3.2. MNR Affects DNA Methylation and Alters Cellular Function, as Shown by KEGG Pathway Analysis
3.3. MNR Regulates the Expression of Genes Associated with the Stress Response and Cellular Defense
3.4. Metabolic Pathways Associated with Immune Function
3.5. The Reprogramming of the Mitochondrial Metabolism Is Associated with Immune Competence
4. Materials and Methods
4.1. Experimental Animals and Diets
4.2. Thymus Sample Collection
4.3. DNA Methylation Analyses
4.3.1. Whole-Genome Bisulfite Sequencing and Library Preparation
4.3.2. Whole-Genome Bisulfite Sequencing Data Processing
4.3.3. Analyses of DMRs and DMGs
4.4. RNA Sequencing Analysis
4.4.1. Sample Preparation for RNA-Seq Analysis
4.4.2. Quantification of Gene Expression and Differential Expression Analysis
4.5. Gene Overlap Analysis
4.6. qPCR Analysis
4.7. Gene Functional Enrichment Analysis
4.8. Metabolome Analysis
4.8.1. Sample Preparation for CE–FTMS Analysis
4.8.2. CE–FTMS Analysis
4.8.3. Metabolomic Data Analysis
4.9. Hierarchical Clustering Analysis (Graphic Visualization)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barker, D.J.; Osmond, C.; Law, C. The intrauterine and early postnatal origins of cardiovascular disease and chronic bronchitis. J. Epidemiol. Community Health 1989, 43, 237–240. [Google Scholar] [CrossRef]
- Matsas, A.; Panopoulou, P.; Antoniou, N.; Bargiota, A.; Gryparis, A.; Vrachnis, N.; Mastorakos, G.; Kalantaridou, S.N.; Panoskaltsis, T.; Vlahos, N.F. Chronic stress in pregnancy is associated with low birth weight: A meta-analysis. J. Clin. Med. 2023, 12, 7686. [Google Scholar] [CrossRef] [PubMed]
- Lowensohn, R.I.; Stadler, D.D.; Naze, C. Current concepts of maternal nutrition. Obstet. Gynecol. Surv. 2016, 71, 413–426. [Google Scholar] [CrossRef]
- Vonnahme, K.A.; Hess, B.W.; Hansen, T.R.; McCormick, R.J.; Rule, D.C.; Moss, G.E.; Murdoch, W.J.; Nijland, M.J.; Skinner, D.C.; Nathanielsz, P.W. Maternal undernutrition from early-to mid-gestation leads to growth retardation, cardiac ventricular hypertrophy, and increased liver weight in the fetal sheep. Biol. Reprod. 2003, 69, 133–140. [Google Scholar] [CrossRef]
- Phomvisith, O.; Takahashi, H.; Mai, H.T.; Shiotsuka, Y.; Matsubara, A.; Sugino, T.; Mcmahon, C.D.; Etoh, T.; Fujino, R.; Furuse, M.; et al. Effects of nutritional status on hormone concentrations of the somatotropin axis and metabolites in plasma and colostrum of japanese black cows. Anim. Sci. J. 2017, 88, 643–652. [Google Scholar] [CrossRef]
- Zhang, Y.; Otomaru, K.; Oshima, K.; Goto, Y.; Oshima, I.; Muroya, S.; Sano, M.; Saneshima, R.; Nagao, Y.; Kinoshita, A.; et al. Effects of low and high levels of maternal nutrition consumed for the entirety of gestation on the development of muscle, adipose tissue, bone, and the organs of wagyu cattle fetuses. Anim. Sci. J. 2021, 92, e13600. [Google Scholar] [CrossRef]
- Muroya, S.; Zhang, Y.; Kinoshita, A.; Otomaru, K.; Oshima, K.; Gotoh, Y.; Oshima, I.; Sano, M.; Roh, S.; Oe, M.; et al. Maternal undernutrition during pregnancy alters amino acid metabolism and gene expression associated with energy metabolism and angiogenesis in fetal calf muscle. Metabolites 2021, 11, 582. [Google Scholar] [CrossRef]
- Muroya, S.; Otomaru, K.; Oshima, K.; Oshima, I.; Ojima, K.; Gotoh, T. DNA methylation of genes participating in hepatic metabolisms and function in fetal calf liver is altered by maternal undernutrition during gestation. Int. J. Mol. Sci. 2023, 24, 10682. [Google Scholar] [CrossRef]
- Armengaud, J.-B.; Dennebouy, Z.; Labes, D.; Fumey, C.; Wilson, A.; Candotti, F.; Yzydorczyk, C.; Simeoni, U. Intra-uterine growth restriction induced by maternal low-protein diet causes long-term alterations of thymic structure and function in adult male rat offspring. Br. J. Nutr. 2020, 123, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Zi, Y.; Ma, C.; He, S.; Yang, H.; Zhang, M.; Gao, F.; Liu, Y. Effects of intrauterine growth restriction during late pregnancy on the cell growth, proliferation, and differentiation in ovine fetal thymuses. Anim. Biosci. 2022, 35, 989. [Google Scholar] [CrossRef]
- Zhou, A.; Ryan, J. Biological embedding of early-life adversity and a scoping review of the evidence for intergenerational epigenetic transmission of stress and trauma in humans. Genes 2023, 14, 1639. [Google Scholar] [CrossRef] [PubMed]
- Novo, N.; Romero-Tamayo, S.; Marcuello, C.; Boneta, S.; Blasco-Machin, I.; Velázquez-Campoy, A.; Villanueva, R.; Moreno-Loshuertos, R.; Lostao, A.; Medina, M. Beyond a platform protein for the degradosome assembly: The apoptosis-inducing factor as an efficient nuclease involved in chromatinolysis. PNAS Nexus 2023, 2, pgac312. [Google Scholar] [CrossRef]
- O’Neill, H.; Lee, H.; Gupta, I.; Rodger, E.J.; Chatterjee, A. Single-cell DNA methylation analysis in cancer. Cancers 2022, 14, 6171. [Google Scholar] [CrossRef]
- Li, Y.; Carrillo, J.A.; Ding, Y.; He, Y.; Zhao, C.; Liu, J.; Zan, L.; Song, J. DNA methylation, microrna expression profiles and their relationships with transcriptome in grass-fed and grain-fed angus cattle rumen tissue. PLoS ONE 2019, 14, e0214559. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, J.; Nagib, P.; Verinaud, L. The thymus microenvironment in regulating thymocyte differentiation. Cell Adhes. Migr. 2010, 4, 382–390. [Google Scholar] [CrossRef]
- Camaglia, F.; Ryvkin, A.; Greenstein, E.; Reich-Zeliger, S.; Chain, B.; Mora, T.; Walczak, A.M.; Friedman, N. Quantifying changes in the t cell receptor repertoire during thymic development. eLife 2023, 12, e81622. [Google Scholar] [CrossRef]
- National Agriculture and Food Research Organization. Japanese Feeding Standard for Beef Cattle 2008 edn; Japan Livestock Industry Association: Tokyo, Japan, 2009. (In Japanese) [Google Scholar]
- Wakitani, S.; Kawabata, R.; Yasuda, M. Insufficiency of cd205-positive cortical thymic epithelial cells in immature japanese black cattle with severe thymic abnormalities and poor prognosis. Vet. Immunol. Immunopathol. 2022, 245, 110379. [Google Scholar] [CrossRef]
- Knapek, K.J.; Georges, H.M.; Van Campen, H.; Bishop, J.V.; Bielefeldt-Ohmann, H.; Smirnova, N.P.; Hansen, T.R. Fetal lymphoid organ immune responses to transient and persistent infection with bovine viral diarrhea virus. Viruses 2020, 12, 816. [Google Scholar] [CrossRef] [PubMed]
- Grandin, T. Welfare problems in cattle, pigs, and sheep that persist even though scientific research clearly shows how to prevent them. Animals 2018, 8, 124. [Google Scholar] [CrossRef]
- Wu, G.; Bazer, F.W.; Cudd, T.A.; Meininger, C.J.; Spencer, T.E. Maternal nutrition and fetal development. J. Nutr. 2004, 134, 2169–2172. [Google Scholar] [CrossRef] [PubMed]
- Takeno, M.A.; Nomura, R.M.Y. Fetal thymus in growth-restricted fetuses due to placental insufficiency. Rev. Assoc. MÉDica Bras. 2023, 69, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Gotardo, A.T.; Dipe, V.V.; Hueza, I.M.; Górniak, S.L. Maternal feed restriction during pregnancy in wistar rats: Evaluation of offspring using classical and immunoteratology protocols. Hum. Exp. Toxicol. 2017, 36, 603–615. [Google Scholar] [CrossRef]
- Sohlstrom, A.; Katsman, A.; Kind, K.; Roberts, C.; Owens, P.; Robinson, J.; Owens, J. Food restriction alters pregnancy-associated changes in igf and igfbp in the guinea pig. Am. J. Physiol.-Endocrinol. Metab. 1998, 274, E410–E416. [Google Scholar] [CrossRef] [PubMed]
- Osgerby, J.; Wathes, D.; Howard, D.; Gadd, T. The effect of maternal undernutrition on ovine fetal growth. J. Endocrinol. 2002, 173, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.E.; Jiang, Z.C.; Alqahtani, F.; Mandoiu, I.; Dong, H.; Zheng, X.; Marjani, S.L.; Chen, J.; Tian, X.C. Methylome dynamics of bovine gametes and in vivo early embryos. Front. Genet. 2019, 10, 512. [Google Scholar] [CrossRef]
- de Mendoza, A.; Nguyen, T.V.; Ford, E.; Poppe, D.; Buckberry, S.; Pflueger, J.; Grimmer, M.R.; Stolzenburg, S.; Bogdanovic, O.; Oshlack, A. Large-scale manipulation of promoter DNA methylation reveals context-specific transcriptional responses and stability. Genome Biol. 2022, 23, 163. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, M.C.; Dickerson, D.R.; Schmitt, M.; Groudine, M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 2004, 11, 1068–1075. [Google Scholar] [CrossRef]
- Tekola-Ayele, F.; Zeng, X.; Ouidir, M.; Workalemahu, T.; Zhang, C.; Delahaye, F.; Wapner, R. DNA methylation loci in placenta associated with birthweight and expression of genes relevant for early development and adult diseases. Clin. Epigenet. 2020, 12, 78. [Google Scholar] [CrossRef]
- Shahoha, M.; Cohen, R.; Ben-Simon, Y.; Ashery, U. Camp-dependent synaptic plasticity at the hippocampal mossy fiber terminal. Front. Synaptic Neurosci. 2022, 14, 861215. [Google Scholar] [CrossRef]
- Jun, J.E.; Rubio, I.; Roose, J.P. Regulation of ras exchange factors and cellular localization of ras activation by lipid messengers in t cells. Front. Immunol. 2013, 4, 57883. [Google Scholar] [CrossRef]
- Iborra, S.; Soto, M.; Stark-Aroeira, L.; Castellano, E.; Alarcón, B.; Alonso, C.; Santos, E.; Fernández-Malavé, E. H-ras and n-ras are dispensable for t-cell development and activation but critical for protective th1 immunity. Blood J. Am. Soc. Hematol. 2011, 117, 5102–5111. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Tolone, A.; Hilhorst, R.; Groten, J.; Tomar, T.; Paquet-Durand, F. Kinase activity profiling identifies putative downstream targets of cgmp/pkg signaling in inherited retinal neurodegeneration. Cell Death Discov. 2022, 8, 93. [Google Scholar] [CrossRef]
- Padgett, D.A.; Glaser, R. How stress influences the immune response. Trends Immunol. 2003, 24, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Swaney, K.F.; Li, R. Function and regulation of the arp2/3 complex during cell migration in diverse environments. Curr. Opin. Cell Biol. 2016, 42, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lu, N.; Xie, C. The hippo and wnt signalling pathways: Crosstalk during neoplastic progression in gastrointestinal tissue. FEBS J. 2019, 286, 3745–3756. [Google Scholar] [CrossRef]
- Ali, D.S.; Dandurand, K.; Khan, A.A. Hypoparathyroidism in pregnancy and lactation: Current approach to diagnosis and management. J. Clin. Med. 2021, 10, 1378. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Wang, T.T.; Ravetch, J.V. The role and function of fcγ receptors on myeloid cells. Myeloid Cells Health Dis. A Synth. 2017, 405–427. [Google Scholar] [CrossRef]
- Sugiura, A.; Beier, K.L.; Chi, C.; Heintzman, D.R.; Ye, X.; Wolf, M.M.; Patterson, A.R.; Cephus, J.-Y.; Hong, H.S.; Lyssiotis, C.A. Tissue-specific dependence of th1 cells on the amino acid transporter slc38a1 in inflammation. bioRxiv 2023. [Google Scholar] [CrossRef]
- Vincenzi, A.; Goettert, M.I.; de Souza, C.F.V. An evaluation of the effects of probiotics on tumoral necrosis factor (tnf-α) signaling and gene expression. Cytokine Growth Factor Rev. 2021, 57, 27–38. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front. Immunol. 2017, 8, 290728. [Google Scholar] [CrossRef]
- Drouin, M.; Saenz, J.; Chiffoleau, E. C-type lectin-like receptors: Head or tail in cell death immunity. Front. Immunol. 2020, 11, 494473. [Google Scholar] [CrossRef]
- Arnold, P.K.; Finley, L.W. Regulation and function of the mammalian tricarboxylic acid cycle. J. Biol. Chem. 2023, 299, 102838. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Distelmaier, F.; Smeitink, J.A.; Willems, P.H. Oxphos mutations and neurodegeneration. EMBO J. 2013, 32, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, Y.; Yuan, S.; Sun, L. Targeting mitochondrial metabolic reprogramming as a potential approach for cancer therapy. Int. J. Mol. Sci. 2023, 24, 4954. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.; Akinloye, O. First line defence antioxidants-superoxide dismutase (sod), catalase (cat) and glutathione peroxidase (gpx): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Ghosh, S. Sialic acid and biology of life: An introduction. In Sialic Acids and Sialoglycoconjugates in the Biology of Life, Health and Disease; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Holeček, M. Histidine in health and disease: Metabolism, physiological importance, and use as a supplement. Nutrients 2020, 12, 848. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-carbon metabolism in health and disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Tian, S.; Feng, J.; Cao, Y.; Shen, S.; Cai, Y.; Yang, D.; Yan, R.; Wang, L.; Zhang, H.; Zhong, X. Glycine cleavage system determines the fate of pluripotent stem cells via the regulation of senescence and epigenetic modifications. Life Sci. Alliance 2019, 2, e201900413. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, Y.; Sun, X.; Zhang, T.; Xu, L.; Xie, H.; Li, Z.; Liu, W.; Lou, J.; Chen, W. Fcγriib-i232t polymorphic change allosterically suppresses ligand binding. eLife 2019, 8, e46689. [Google Scholar] [CrossRef] [PubMed]
- Miri, S.; Yeo, J.; Abubaker, S.; Hammami, R. Neuromicrobiology, an emerging neurometabolic facet of the gut microbiome? Front. Microbiol. 2023, 14, 1098412. [Google Scholar] [CrossRef]
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-tryptophan: Basic metabolic functions, behavioral research and therapeutic indications. Int. J. Tryptophan Res. 2009, 2, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, A.-H.; Song, Q.; Fang, H.; Liu, X.-Y.; Su, J.; Yang, L.; Yu, M.-D.; Wang, X.-J. Functional metabolomics discover pentose and glucuronate interconversion pathways as promising targets for yang huang syndrome treatment with yinchenhao tang. RSC Adv. 2018, 8, 36831–36839. [Google Scholar] [CrossRef]
- Prentki, M.; Madiraju, S.M. Glycerolipid metabolism and signaling in health and disease. Endocr. Rev. 2008, 29, 647–676. [Google Scholar] [CrossRef]
- Gimenez, M.S.; Oliveros, L.B.; Gomez, N.N. Nutritional deficiencies and phospholipid metabolism. Int. J. Mol. Sci. 2011, 12, 2408–2433. [Google Scholar] [CrossRef] [PubMed]
- Beyoğlu, D.; Huang, P.; Skelton-Badlani, D.; Zong, C.; Popov, Y.V.; Idle, J.R. Metabolic hijacking of hexose metabolism to ascorbate synthesis is the unifying biochemical basis of murine liver fibrosis. Cells 2023, 12, 485. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Chen, L.; Wan, J.; Liu, W.; Lou, S.; Shen, Z. Single-cell transcriptomic landscape of immunometabolism reveals intervention candidates of ascorbate and aldarate metabolism, fatty-acid degradation and pufa metabolism of t-cell subsets in healthy controls, psoriasis and psoriatic arthritis. Front. Immunol. 2023, 14, 1179877. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Huang, Z.; Xiong, W.; Ye, H.; Deng, J.; Yin, Y. A review of the amino acid metabolism in placental function response to fetal loss and low birth weight in pigs. J. Anim. Sci. Biotechnol. 2022, 13, 28. [Google Scholar] [CrossRef]
- Kwon, H.; Ford, S.P.; Bazer, F.W.; Spencer, T.E.; Nathanielsz, P.W.; Nijland, M.J.; Hess, B.W.; Wu, G. Maternal nutrient restriction reduces concentrations of amino acids and polyamines in ovine maternal and fetal plasma and fetal fluids. Biol. Reprod. 2004, 71, 901–908. [Google Scholar] [CrossRef]
- Muroya, S.; Zhang, Y.; Otomaru, K.; Oshima, K.; Oshima, I.; Sano, M.; Roh, S.; Ojima, K.; Gotoh, T. Maternal nutrient restriction disrupts gene expression and metabolites associated with urea cycle, steroid synthesis, glucose homeostasis, and glucuronidation in fetal calf liver. Metabolites 2022, 12, 203. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Angeles-Valencia, M.; Morales-González, Á.; Madrigal-Santillán, E.O.; Morales-Martínez, M.; Madrigal-Bujaidar, E.; Álvarez-González, I.; Gutiérrez-Salinas, J.; Esquivel-Chirino, C.; Chamorro-Cevallos, G. Oxidative stress, mitochondrial function and adaptation to exercise: New perspectives in nutrition. Life 2021, 11, 1269. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial tca cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Sokolova, I. Mitochondrial adaptations to variable environments and their role in animals’ stress tolerance. Integr. Comp. Biol. 2018, 58, 519–531. [Google Scholar] [CrossRef]
- Thompson, K.; Majd, H.; Dallabona, C.; Reinson, K.; King, M.S.; Alston, C.L.; He, L.; Lodi, T.; Jones, S.A.; Fattal-Valevski, A. Recurrent de novo dominant mutations in slc25a4 cause severe early-onset mitochondrial disease and loss of mitochondrial DNA copy number. Am. J. Hum. Genet. 2016, 99, 860–876. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.-W.; Jiang, K.; Shen, X.-W.; Ma, Z.-R.; Yan, X.-M.; Xia, H.; Cao, X. The requirement of the mitochondrial protein ndufs8 for angiogenesis. Cell Death Dis. 2024, 15, 253. [Google Scholar] [CrossRef]
- Gardeitchik, T.; Mohamed, M.; Ruzzenente, B.; Karall, D.; Guerrero-Castillo, S.; Dalloyaux, D.; van den Brand, M.; van Kraaij, S.; van Asbeck, E.; Assouline, Z. Bi-allelic mutations in the mitochondrial ribosomal protein mrps2 cause sensorineural hearing loss, hypoglycemia, and multiple oxphos complex deficiencies. Am. J. Hum. Genet. 2018, 102, 685–695. [Google Scholar] [CrossRef] [PubMed]
- McCammon, M.T.; Epstein, C.B.; Przybyla-Zawislak, B.; McAlister-Henn, L.; Butow, R.A. Global transcription analysis of krebs tricarboxylic acid cycle mutants reveals an alternating pattern of gene expression and effects on hypoxic and oxidative genes. Mol. Biol. Cell 2003, 14, 958–972. [Google Scholar] [CrossRef]
- Cappel, D.A.; Deja, S.; Duarte, J.A.; Kucejova, B.; Iñigo, M.; Fletcher, J.A.; Fu, X.; Berglund, E.D.; Liu, T.; Elmquist, J.K. Pyruvate-carboxylase-mediated anaplerosis promotes antioxidant capacity by sustaining tca cycle and redox metabolism in liver. Cell Metab. 2019, 29, 1291–1305.e1298. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; Mayer, S.; Thiel, G.; Mercy, L.; Arnould, T.; Hornig-Do, H.-T.; Wiesner, R.J.; Goffart, S. Creb-1α is recruited to and mediates upregulation of the cytochrome c promoter during enhanced mitochondrial biogenesis accompanying skeletal muscle differentiation. Mol. Cell. Biol. 2008, 28, 2446–2459. [Google Scholar] [CrossRef]
- Tubert, C.; Zampese, E.; Pancani, T.; Tkatch, T.; Surmeier, D.J. Feed-forward metabotropic signaling by cav1 ca2+ channels supports pacemaking in pedunculopontine cholinergic neurons. Neurobiol. Dis. 2023, 188, 106328. [Google Scholar] [CrossRef]
- Kim, J.-E.; Lee, D.-S.; Park, H.; Kim, T.-H.; Kang, T.-C. Inhibition of akt/gsk3β/creb pathway improves the responsiveness to ampa receptor antagonists by regulating gria1 surface expression in chronic epilepsy rats. Biomedicines 2021, 9, 425. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Martin, P.M.; Chen, W. Mitochondrial oxidative stress and energy metabolism: Impact on aging and longevity. Oxidative Med. Cell. Longev. 2021, 2021, 9789086. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Wilcox, W.R.; Cederbaum, S.D. Amino acid metabolism. Emery Rimoin’S Essent. Med. Genet. 2013, 372, 1–42. [Google Scholar] [CrossRef]
- Jochmanova, I.; Pacak, K. Pheochromocytoma: The first metabolic endocrine cancer. Clin. Cancer Res. 2016, 22, 5001–5011. [Google Scholar] [CrossRef]
- Hara, Y.; Kume, S.; Kataoka, Y.; Watanabe, N. Changes in tca cycle and tca cycle-related metabolites in plasma upon citric acid administration in rats. Heliyon 2021, 7, e08501. [Google Scholar] [CrossRef]
- Guda, P.; Guda, C.; Subramaniam, S. Reconstruction of pathways associated with amino acid metabolism in human mitochondria. Genom. Proteom. Bioinform. 2007, 5, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Yiew, N.K.; Vazquez, J.H.; Martino, M.R.; Kennon-McGill, S.; Price, J.R.; Allard, F.D.; Yee, E.U.; Layman, A.J.; James, L.P.; McCommis, K.S. Hepatic pyruvate and alanine metabolism are critical and complementary for maintenance of antioxidant capacity and resistance to oxidative insult. Mol. Metab. 2023, 77, 101808. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, J.H.F.; Esteves-Ferreira, A.A.; Quinhones, C.G.; Pereira-Lima, I.A.; Nunes-Nesi, A.; Fernie, A.R.; Araujo, W.L. Evolution and functional implications of the tricarboxylic acid cycle as revealed by phylogenetic analysis. Genome Biol. Evol. 2014, 6, 2830–2848. [Google Scholar] [CrossRef]
- Tang, Q.; Tan, P.; Ma, N.; Ma, X. Physiological functions of threonine in animals: Beyond nutrition metabolism. Nutrients 2021, 13, 2592. [Google Scholar] [CrossRef]
- Marquez, S.; Fernández, J.J.; Mancebo, C.; Herrero-Sanchez, C.; Alonso, S.; Sandoval, T.A.; Prados, M.R.; Cubillos-Ruiz, J.R.; Montero, O.; Fernandez, N. Tricarboxylic acid cycle activity and remodeling of glycerophosphocholine lipids support cytokine induction in response to fungal patterns. Cell Rep. 2019, 27, 525–536.e524. [Google Scholar] [CrossRef]
- Liu, N.; Yan, M.; Tao, Q.; Wu, J.; Chen, J.; Chen, X.; Peng, C. Inhibition of tca cycle improves the anti-pd-1 immunotherapy efficacy in melanoma cells via atf3-mediated pd-l1 expression and glycolysis. J. Immunother. Cancer 2023, 11, e007146. [Google Scholar] [CrossRef]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor creb in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Son, H.; Baek, J.-H. Tricarboxylic acid (tca) cycle intermediates: Regulators of immune responses. Life 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of cd4+ t cells in cd8+ t cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Pei, X.-Y.; Shen, H.; Gao, Y.-N.; Sun, X.-Y.; Wang, W.; Ge, Q.; Zhang, Y. Thymic homing of activated cd4+ t cells induces degeneration of the thymic epithelium through excessive rank signaling. Sci. Rep. 2017, 7, 2421. [Google Scholar] [CrossRef]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The sanger fastq file format for sequences with quality scores, and the solexa/illumina fastq variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.D.; He, Y.; Whitaker, J.W.; Hariharan, M.; Mukamel, E.A.; Leung, D.; Rajagopal, N.; Nery, J.R.; Urich, M.A.; Chen, H. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 2015, 523, 212–216. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. Methylkit: A comprehensive r package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef]
- Schaarschmidt, S.; Fischer, A.; Zuther, E.; Hincha, D.K. Evaluation of seven different rna-seq alignment tools based on experimental data from the model plant arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 1720. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of rna-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Barber, R.F. Multiple testing with the structure-adaptive benjamini–hochberg algorithm. J. R. Stat. Soc. Ser. B Stat. Methodol. 2019, 81, 45–74. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2− δδct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. Shinygo: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Volzhenin, K.; Bittner, L.; Carbone, A. Sense-ppi reconstructs protein-protein interactions of various complexities, within, across, and between species, with sequence-based evolutionary scale modeling and deep learning. bioRxiv 2023. [Google Scholar] [CrossRef]
- Arif, M.; Basu, A.; Wolf, K.M.; Park, J.K.; Pommerolle, L.; Behee, M.; Gochuico, B.R.; Cinar, R. An integrative multiomics framework for identification of therapeutic targets in pulmonary fibrosis. Adv. Sci. 2023, 10, e2207454. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. Srplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component. | HN (n = 6) | LN (n = 5) | Ratio | |||
|---|---|---|---|---|---|---|
| Mean | SEM 1 | Mean | SEM 1 | LN/HN | p-Value | |
| Fetal age (d) | 261.2 | 1.3 | 261.6 | 1.6 | 1.0 | 0.835 |
| Fetal weight (g) | 32,653.3 | 448.5 | 23,976.0 | 2540.9 | 0.7 | 0.005 |
| Thymus (g) | 212.0 | 21.2 | 110.5 | 22.2 | 0.5 | 0.009 |
| % Body weight | 0.7 | 0.001 | 0.4 | 0.001 | 0.7 | 0.046 |
| Genomic Regions | Hypo | Hyper | Ratio (Hypo/Hyper) | ||
|---|---|---|---|---|---|
| Count | % | Count | % | ||
| Promoter | 63 | 0.9 | 111 | 1.7 | 0.6 |
| 1–5 kb | 278 | 3.8 | 253 | 3.9 | 1.1 |
| Exon | 55 | 0.8 | 76 | 1.2 | 0.7 |
| Intergenic | 3876 | 52.9 | 3317 | 51.2 | 1.2 |
| Intron | 2220 | 30.3 | 1891 | 29.2 | 1.2 |
| Intron-exon boundary | 764 | 10.4 | 765 | 11.8 | 1.0 |
| None | 65 | 0.9 | 62 | 1.0 | 1.0 |
| Total | 7321 | 100.0 | 6475 | 100.0 | |
| Methylated Genes | KEGG Pathway | q-Value |
|---|---|---|
| Hypo | ||
| Path:bta04720 Long-term potentiation | 0.00007 | |
| Path:bta04360 Axon guidance | 0.00010 | |
| Path:bta04924 Renin secretion | 0.00015 | |
| Path:bta04072 Phospholipase D signaling pathway | 0.00015 | |
| Path:bta04927 Cortisol synthesis and secretion | 0.00015 | |
| Path:bta04921 Oxytocin signaling pathway | 0.00018 | |
| Path:bta04925 Aldosterone synthesis and secretion | 0.00018 | |
| Path:bta04971 Gastric acid secretion | 0.00027 | |
| Path:bta04540 Gap junction | 0.00035 | |
| Path:bta04912 GnRH signaling pathway | 0.00035 | |
| Path:bta04916 Melanogenesis | 0.00038 | |
| Path:bta04935 Growth hormone synthesis secretion and action | 0.00039 | |
| Path:bta04970 Salivary secretion | 0.00039 | |
| Path:bta05207 Chemical carcinogenesis-receptor activation | 0.00039 | |
| Path:bta05033 Nicotine addiction | 0.00039 | |
| Path:bta05414 Dilated cardiomyopathy | 0.00039 | |
| Path:bta04810 Regulation of actin cytoskeleton | 0.00040 | |
| Path:bta04730 Long-term depression | 0.00041 | |
| Path:bta04014 Ras signaling pathway | 0.00050 | |
| Hyper | ||
| Path:bta04926 Relaxin signaling pathway | 0.00081 | |
| Path:bta04666 Fc gamma R-mediated phagocytosis | 0.00070 | |
| Path:bta05032 Morphine addiction | 0.00070 | |
| Path:bta05231 Choline metabolism in cancer | 0.00070 | |
| Path:bta04310 Wnt signaling pathway | 0.00096 |
| Direction | KEGG Pathway | q-Value | Genes |
|---|---|---|---|
| Upregulated | |||
| Path:bta03010 Ribosome | <0.0001 | RPS14, RPLP2, RPL37A, MRPS2, RPS15, RPLP1, RPS21, UBA52, RPL38, RPL28, RPL37 | |
| Path:bta05171 Coronavirus disease-COVID-19 | <0.0001 | RPS14, RPLP2, RPL37A, RPS15, RPLP1, RPS21, UBA52, RPL38, RPL28, RPL37 | |
| Path:bta05022 Pathways of neurodegeneration—multiple diseases | 0.002 | GPX1, SLC25A4, NDUFS8, NDUFB10, CACNA1D, CYC1, GRIA1, UBA52 | |
| Path:bta05016 Huntington disease | 0.005 | GPX1, SLC25A4, NDUFS8, NDUFB10, CYC1, GRIA1 | |
| Path:bta00480 Glutathione metabolism | 0.010 | GPX1, GPX4, GGT7 | |
| Path:bta04723 Retrograde endocannabinoid signaling | 0.011 | NDUFS8, NDUFB10, CACNA1D, GRIA1 | |
| Path:bta04932 Non-alcoholic fatty liver disease | 0.011 | NDUFS8, NDUFB10, CYC1, SREBF1 | |
| Path:bta05012 Parkinson disease | 0.011 | SLC25A4, NDUFS8, NDUFB10, CYC1, UBA52 | |
| Path:bta05020 Prion disease | 0.011 | SLC25A4, NDUFS8, NDUFB10, CACNA1D, CYC1 | |
| Path:bta05415 Diabetic cardiomyopathy | 0.026 | SLC25A4, NDUFS8, NDUFB10, CYC1 | |
| Path:bta05014 Amyotrophic lateral sclerosis | 0.029 | GPX1, NDUFS8, NDUFB10, CYC1, GRIA1 | |
| Path:bta05208 Chemical carcinogenesis-reactive oxygen species | 0.032 | SLC25A4, NDUFS8, NDUFB10, CYC1 | |
| Path:bta05010 Alzheimer disease | 0.032 | SLC25A4, NDUFS8, NDUFB10, CACNA1D, CYC1 | |
| Path:bta00190 Oxidative phosphorylation | 0.047 | NDUFS8, NDUFB10, CYC1 | |
| Path:bta01100 Metabolic pathways | 0.064 | MIF, GPX1, GPX4, NDUFS8, NDUFB10, PC, IDUA, ACAD8, CYC1, GGT7 | |
| Path:bta05031 Amphetamine addiction | 0.078 | CACNA1D, GRIA1 | |
| Downregulated | |||
| Path:bta04010 MAPK signaling pathway | 0.077 | NLK, MAP3K20, PAK1 | |
| Path:bta04625 C-type lectin receptor signaling pathway | 0.077 | PTPN11, PAK1 | |
| Path:bta05211 Renal cell carcinoma | 0.077 | PTPN11, PAK1 | |
| Path:bta04650 Natural killer cell-mediated cytotoxicity | 0.094 | PTPN11, PAK1 |
| Compound Name | HN (n = 6) | LN (n = 5) | LN/HN | |||
|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | p-Value | Ratio | |
| UDP-glucose, UDP-galactose | 0.01845 | 0.00148 | 0.01190 | 0.000449 | 0.004 | 0.6 |
| 2-Deoxyadenosine | 0.00001 | 0.00000 | 0.00002 | 0.000001 | 0.010 | 1.3 |
| Ethanolamine phosphate | 0.22594 | 0.00462 | 0.24262 | 0.002295 | 0.014 | 1.1 |
| His | 0.03384 | 0.00152 | 0.04124 | 0.002097 | 0.017 | 1.2 |
| N-acetylmethionine | 0.00172 | 0.00010 | 0.00137 | 0.000074 | 0.020 | 0.8 |
| Ribulose 5-phosphate | 0.00150 | 0.00011 | 0.00203 | 0.000173 | 0.023 | 1.4 |
| 3-(3,4-Dihydroxyphenyl)-2-methylalanine | 0.00013 | 0.00002 | 0.00024 | 0.000038 | 0.023 | 1.9 |
| Ala-Ala | 0.00152 | 0.00010 | 0.00188 | 0.000092 | 0.028 | 1.2 |
| Ser | 0.05118 | 0.00501 | 0.07142 | 0.006726 | 0.036 | 1.4 |
| Histamine | 0.00112 | 0.00018 | 0.00169 | 0.000143 | 0.039 | 1.5 |
| Gluconolactone | 0.00050 | 0.00003 | 0.00063 | 0.000046 | 0.044 | 1.2 |
| 5-Hydroxylysine | 0.00099 | 0.00006 | 0.00124 | 0.000084 | 0.044 | 1.2 |
| 1H-Imidazole-4-propionic acid | 0.00004 | 0.00001 | 0.00013 | 0.000041 | 0.047 | 2.9 |
| N-acetylglucosamine 1-phosphate | 0.00375 | 0.00010 | 0.00465 | 0.000424 | 0.050 | 1.2 |
| Phe | 0.12468 | 0.00845 | 0.15371 | 0.009901 | 0.051 | 1.2 |
| Terephthalic acid | 0.00016 | 0.00001 | 0.00011 | 0.000017 | 0.054 | 0.7 |
| dCMP | 0.00153 | 0.00007 | 0.00123 | 0.000124 | 0.054 | 0.8 |
| Nicotinamide | 0.05056 | 0.00397 | 0.06163 | 0.002909 | 0.058 | 1.2 |
| Allo-threonine | 0.000227 | 0.000033 | 0.000367 | 0.000041 | 0.064 | 1.6 |
| Imidazole-4-acetic acid | 0.000016 | 0.000003 | 0.000028 | 0.000005 | 0.064 | 1.7 |
| Pathway | Hits | Total | p-Value | Increased in LN Group | Decreased in LN Group |
|---|---|---|---|---|---|
| Amino sugar and nucleotide sugar metabolism | 6 | 42 | <0.001 | N-Acetylglucosamine 1-phosphate and CMP-N-acetylneuraminic acid | UDP-N-acetylglucosamine; UDP-glucose; UDP-galactose; UDP-N-acetylgalactosamine |
| Histidine metabolism | 4 | 16 | 0.001 | His; Histamine; Imidazole-4-acetic acid; and 1-Methyl-4-imidazoleacetic acid | |
| Glycine, serine, and threonine metabolism | 5 | 34 | 0.001 | Serine; Choline; Allo-threonine; and 4-Guanidinoacetic acid | Cystathionine |
| Phenylalanine, tyrosine, and tryptophan biosynthesis | 2 | 4 | 0.004 | Phenylalanine and tyrosine | |
| Pentose and glucuronate interconversions | 3 | 19 | 0.011 | Ribulose 5-phosphate and Glucuronic acid | UDP-glucose |
| Glycerophospholipid metabolism | 4 | 36 | 0.012 | Choline phosphate; Choline; Ethanolamine phosphate; Ethanolamine | |
| Phenylalanine metabolism | 2 | 10 | 0.025 | Phenylalanine; and Tyrosine | |
| Ascorbate and aldarate metabolism | 2 | 10 | 0.025 | Glucuronic acid | UDP-glucose |
| Tyrosine metabolism | 3 | 42 | 0.089 | Tyrosine; Metanephrine; and Homovanillic acid |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phomvisith, O.; Muroya, S.; Otomaru, K.; Oshima, K.; Oshima, I.; Nishino, D.; Haginouchi, T.; Gotoh, T. Maternal Undernutrition Affects Fetal Thymus DNA Methylation, Gene Expression, and, Thereby, Metabolism and Immunopoiesis in Wagyu (Japanese Black) Cattle. Int. J. Mol. Sci. 2024, 25, 9242. https://doi.org/10.3390/ijms25179242
Phomvisith O, Muroya S, Otomaru K, Oshima K, Oshima I, Nishino D, Haginouchi T, Gotoh T. Maternal Undernutrition Affects Fetal Thymus DNA Methylation, Gene Expression, and, Thereby, Metabolism and Immunopoiesis in Wagyu (Japanese Black) Cattle. International Journal of Molecular Sciences. 2024; 25(17):9242. https://doi.org/10.3390/ijms25179242
Chicago/Turabian StylePhomvisith, Ouanh, Susumu Muroya, Konosuke Otomaru, Kazunaga Oshima, Ichiro Oshima, Daichi Nishino, Taketo Haginouchi, and Takafumi Gotoh. 2024. "Maternal Undernutrition Affects Fetal Thymus DNA Methylation, Gene Expression, and, Thereby, Metabolism and Immunopoiesis in Wagyu (Japanese Black) Cattle" International Journal of Molecular Sciences 25, no. 17: 9242. https://doi.org/10.3390/ijms25179242