
Application of Pan-Omics Technologies in Research on Important Economic Traits for Ruminants


Abstract
:1. Introduction
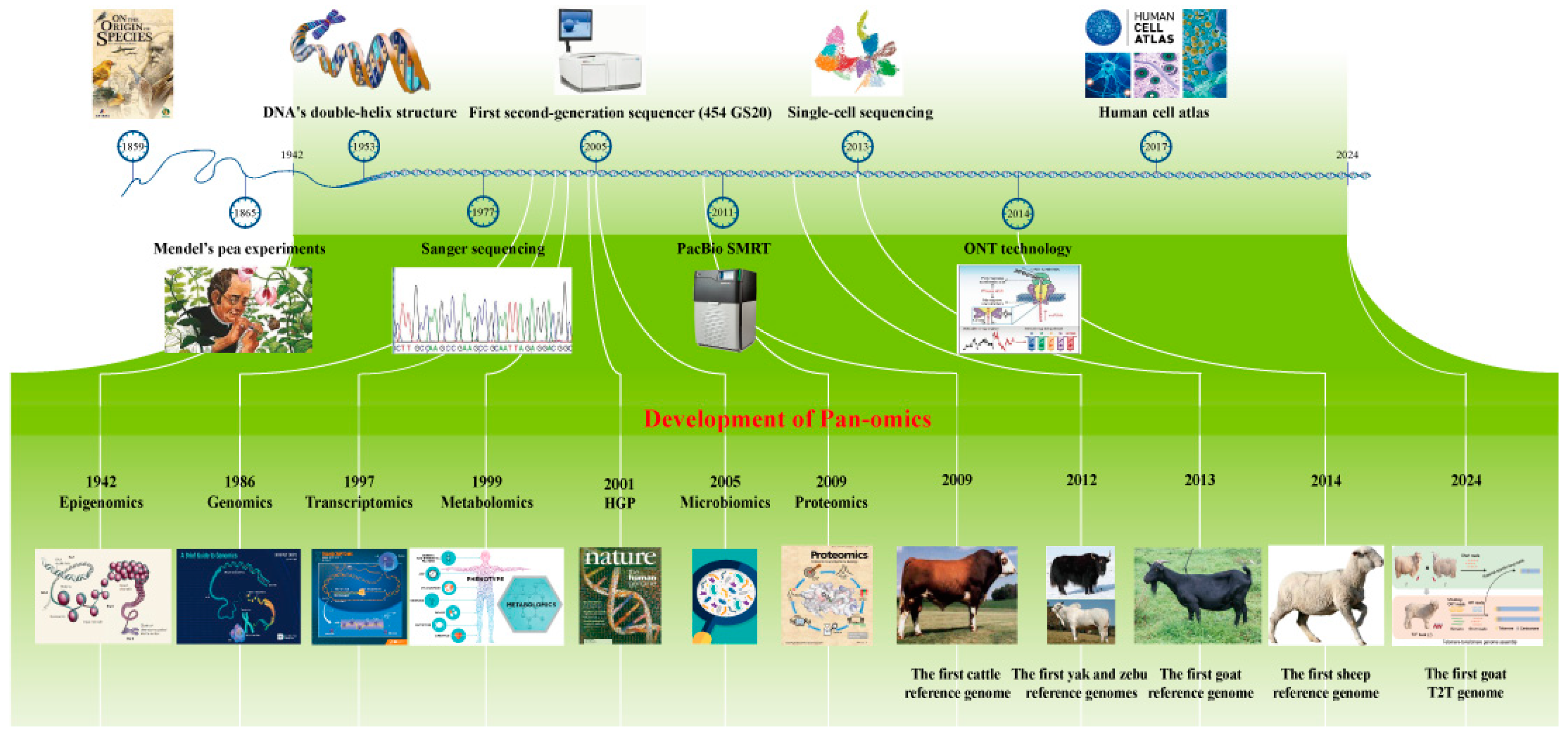
2. Development of Sequencing Technologies for Ruminants
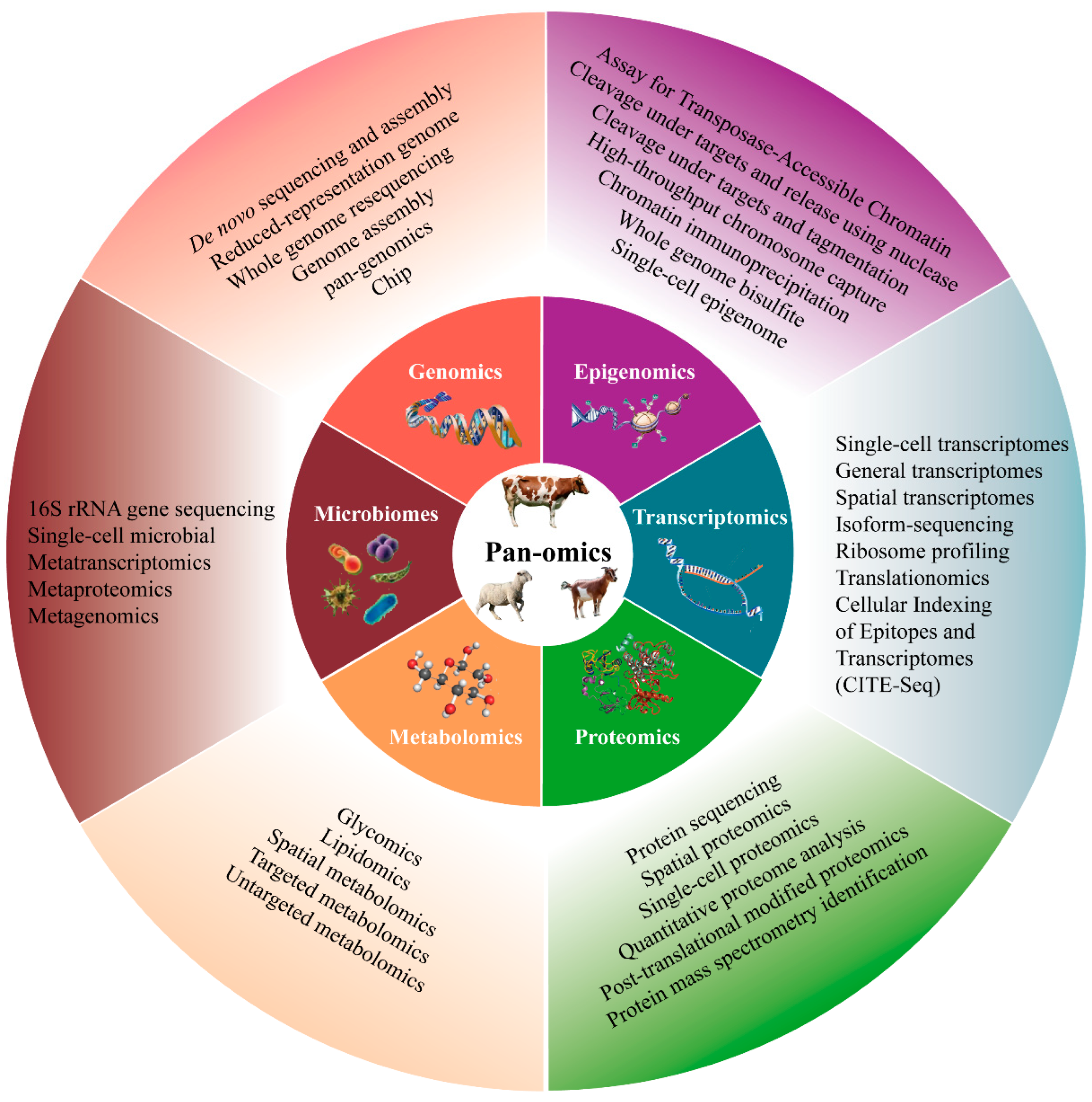
3. Single-Omics Technologies
3.1. Genomics
3.2. Epigenomics
3.3. Transcriptomics
3.4. Proteomics
3.5. Metabolomics
3.6. Microbiomics
4. Pan-Omics Integrated Analysis and Data Integration Methods
5. Application of Pan-Omics Technologies in the Study of Important Economic Traits and Functions of Ruminants
5.1. Application of Pan-Omics Technologies in the Study of Growth and Reproductive Traits of Ruminants
5.2. Application of Pan-Omics Technologies in the Study of Production Performance of Ruminants
5.3. Application of Pan-Omics Technologies in the Study of the Digestive System and Symbiotic Mechanisms of Ruminants
5.4. Application of Pan-Omics Technologies in Adaptation to Extreme Environments and Diseases Prevention in Ruminants
6. Conclusions and Outlook
- (1)
- Data Integration and Interpretation: The integration of multi-omics data (genomics, transcriptomics, proteomics, metabolomics, etc.) poses significant challenges due to the complexity of biological systems and the sheer volume of data generated. The heterogeneity of data types and the lack of standardized analytical frameworks can lead to difficulties in data harmonization and interpretation, ultimately affecting the reliability and reproducibility of findings [130].
- (2)
- Technological and Computational Barriers: Advanced computational tools and resources are required to handle, process, and analyze large-scale omics data. However, the availability of such resources can be limited, particularly in the context of ruminant research, where funding and access to cutting-edge technology may be constrained. Additionally, the development and application of machine learning algorithms for predictive modeling in ruminants are still in their infancy, posing a barrier to the widespread adoption of pan-omics [131].
- (3)
- Biological Complexity and Trait Variability: Ruminants exhibit a high degree of biological complexity and trait variability, influenced by environmental factors, management practices, and genetic diversity. This complexity can obscure the relationships between omics layers and phenotypic traits, making it challenging to draw meaningful conclusions. Furthermore, the dynamic nature of the ruminant microbiome adds another layer of complexity to pan-omics studies [132].
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Loftus, R.T.; Machugh, D.E.; Bradley, D.G.; Sharp, P.M.; Cunningham, P. Evidence for two independent domestications of cattle. Proc. Natl. Acad. Sci. USA 1994, 91, 2757–2761. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, W.R.; Lv, F.H.; He, S.G.; Tian, S.L.; Peng, W.F.; Sun, Y.W.; Zhao, Y.X.; Tu, X.L.; Zhang, M.; et al. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 2016, 33, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Amills, M.; Capote, J.; Tosser-Klopp, G. Goat domestication and breeding: A jigsaw of historical, biological and molecular data with missing pieces. Anim. Genet. 2017, 48, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Darwin, C. On the origin of species by means of natural selection. Am. Anthropol. 1963, 61, 176–177. [Google Scholar] [CrossRef]
- Clarke, C.A. Experiments in plant hybridization. BMJ 1965, 4, 3–47. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H.C. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 248, 623–624. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, J.C.; Hutchison, C.A.; Slocombe, P.M.; Smith, M. Nucleotide sequence of bacteriophage φx174 DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef]
- Gong, Y.; Li, Y.; Liu, X.; Ma, Y.; Jiang, L. A review of the pangenome: How it affects our understanding of genomic variation, selection and breeding in domestic animals? J. Anim. Sci. Biotechnol. 2023, 14, 73. [Google Scholar] [CrossRef]
- Weissenbach, J. The rise of genomics. Comptes Rendus Biol. 2016, 339, 231–239. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, R.A.; Burlingame, A.L. From proteins to proteomics. IUBMB Life 2005, 57, 267–272. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef]
- Whipps, J.; Lewis, K.; Cooke, R. Mycoparasitism and plant disease control. In Fungi in Biological Control Systems; Manchester University Press: Manchester, UK, 1988; pp. 161–187. [Google Scholar]
- Claudia, M.; Kia, D.A.; Jana, V.; John, H.; Wood, N.W.; Lewis, P.A.; Raffaele, F. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Li, M.; Lu, Y.; Gao, Z.; Yue, D.; Hong, J.; Wu, J.; Xi, D.; Deng, W.; Chong, Y. Pan-omics in sheep: Unveiling genetic landscapes. Animals 2024, 14, 273. [Google Scholar] [CrossRef]
- Hackmann, T.J.; Spain, J.N. Invited review: Ruminant ecology and evolution: Perspectives useful to ruminant livestock research and production. J. Dairy Sci. 2010, 93, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Bovine Genome Sequencing and Analysis Consortium; Elsik, C.G.; Tellam, R.L.; Worley, K.C.; Gibbs, R.A.; Muzny, D.M.; Weinstock, G.M.; Adelson, D.L.; Eichler, E.E.; Elnitski, L.; et al. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science 2009, 324, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Zhang, G.J.; Ma, T.; Qian, W.B.; Wang, J.Y.; Ye, Z.Q.; Cao, C.C.; Hu, Q.J.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef]
- Canavez, F.C.; Luche, D.D.; Stothard, P.; Leite, K.R.; Sousa-Canavez, J.M.; Plastow, G.; Meidanis, J.; Souza, M.A.; Feijao, P.; Moore, S.S.; et al. Genome sequence and assembly of bos indicus. J. Hered. 2012, 103, 342–348. [Google Scholar] [CrossRef]
- Dong, Y.; Xie, M.; Jiang, Y.; Xiao, N.; Du, X.; Zhang, W.; Tosser-Klopp, G.; Wang, J.; Yang, S.; Liang, J.; et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 2013, 31, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W.; et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Rosen, B.D.; Koren, S.; Sayre, B.L.; Hastie, A.R.; Chan, S.; Lee, J.; Lam, E.T.; Liachko, I.; Sullivan, S.T.; et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 2017, 49, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gong, M.; Zhang, X.; Wang, F.; Liu, Z.; Zhang, L.; Yang, Q.; Xu, Y.; Xu, M.; Zhang, H.; et al. A sheep pangenome reveals the spectrum of structural variations and their effects on tail phenotypes. Genome Res. 2023, 33, 463–477. [Google Scholar] [CrossRef]
- Wei, Z.C.; Zhang, L.L.; Gao, L.T.; Chen, J.; Peng, L.; Yang, L.N. Chromosome-level genome assembly and annotation of the Yunling cattle with PacBio and hi-c sequencing data. Sci. Data 2024, 11, 233. [Google Scholar] [CrossRef]
- Hood, L. Systems biology: Integrating technology, biology, and computation. Mech. Ageing Dev. 2003, 124, 9–16. [Google Scholar] [CrossRef]
- Venter, J.; Adams, M.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Greely, H.T. Human genome diversity: What about the other human genome project? Nat. Rev. Genet. 2001, 2, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.D.; Lu, Y.; Chong, Y.Q.; Li, M.F.; Hong, J.; Wu, J.; Wu, D.W.; Xi, D.M.; Deng, W.D. Beef cattle genome project: Advances in genome sequencing, assembly, and functional genes discovery. Int. J. Mol. Sci. 2024, 25, 7147. [Google Scholar] [CrossRef]
- Turner, D.J.; Keane, T.M.; Sudbery, I.; Adams, D.J. Next-generation sequencing of vertebrate experimental organisms. Mamm. Genome 2009, 20, 327–338. [Google Scholar] [CrossRef]
- Yang, J.; Wang, D.F.; Huang, J.H.; Zhu, Q.H.; Luo, L.Y.; Lu, R.; Xie, X.L.; Salehian-Dehkordi, H.; Esmailizadeh, A.; Liu, G.E.; et al. Structural variant landscapes reveal convergent signatures of evolution in sheep and goats. Genome Biol. 2024, 25, 148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, Z.; Zhang, M.; Wang, S.; Gao, T.; Huang, H.; Zhang, T.; Cai, H.; Liu, X.; Fu, T.; et al. Population structure and selection signal analysis of Nanyang cattle based on whole-genome sequencing data. Genes 2024, 15, 351. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.; Donati, C.; Medini, D.; Ward, N.; Angiuoli, S.; Crabtree, J.; Jones, A.; Durkin, A.; et al. Genome analysis of multiple pathogenic isolates of streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- A panoply of pangenomes. Nat. Ecol. Evol. 2024, 8, 833. [CrossRef]
- Zhou, Y.; Yang, L.; Han, X.T.; Han, J.Z.; Hu, Y.; Li, F.; Xia, H.; Peng, L.W.; Boschiero, C.; Rosen, B.D.; et al. Assembly of a pangenome for global cattle reveals missing sequences and novel structural variations, providing new insights into their diversity and evolutionary history. Genome. Res. 2022, 32, 1585–1601. [Google Scholar] [CrossRef]
- Milia, S.; Leonard, A.S.; Mapel, X.M.; Ulloa, S.M.B.; Drögemüller, C.; Pausch, H. Taurine Pangenome Uncovers a Segmental Duplication Upstream of Kit Associated with Depigmentation in White-Headed Cattle. bioRxiv 2024. [Google Scholar] [CrossRef]
- Waddington, C.H. The epigenotype 1942. Int. J. Epidemiol. 2012, 41, 10. [Google Scholar] [CrossRef]
- Jablonka, E.; Lamb, M.J. The changing concept of epigenetics. Ann. N. Y. Acad. Sci. 2002, 981, 82–96. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Epigenomics: Technologies and applications. Circ. Res. 2018, 122, 1191–1199. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Lusser, A. Acetylated, methylated, remodeled: Chromatin states for gene regulation. Curr. Opin. Plant Biol. 2002, 5, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Cretney, E.C.; Kropp, J.; Khateeb, K.; Berg, K.K.; Peñagaricano, F.; Magness, R.; Radunz, A.E.; Khatib, H. Maternal diet during pregnancy induces gene expression and DNA methylation changes in fetal tissues in sheep. Front. Genet. 2013, 4, 49. [Google Scholar] [CrossRef]
- Kropp, J.; Carrillo, J.A.; Namous, H.; Daniels, A.; Salih, S.M.; Song, J.; Khatib, H. Male fertility status is associated with DNA methylation signatures in sperm and transcriptomic profiles of bovine preimplantation embryos. BMC Genom. 2017, 18, 280. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, J.; Li, B.; Sun, B.; Yu, S.; Wang, X.; Zan, L. Long non-coding RNA bnip3 inhibited the proliferation of bovine intramuscular preadipocytes via cell cycle. Int. J. Mol. Sci. 2023, 24, 4234. [Google Scholar] [CrossRef]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by polycomb and compass families. Science 2016, 352, aad9780. [Google Scholar] [CrossRef]
- Wang, Y.; Fischle, W.; Cheung, W.; Jacobs, S.; Khorasanizadeh, S.; Allis, C.D. Beyond the double helix: Writing and reading the histone code. Novartis Found Symp. 2004, 259, 3–169. [Google Scholar]
- Velculescu, V.E.; Zhang, L.; Zhou, W.; Vogelstein, J.; Basrai, M.A.; Bassett, D.E.; Hieter, P.; Vogelstein, B.; Kinzler, K.W. Characterization of the yeast transcriptome. Cell 1997, 88, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, D.J.; Dong, H.; Byrne, M.C.; Follettie, M.T.; Gallo, M.V.; Chee, M.S.; Mittmann, M.; Wang, C.; Kobayashi, M.; Norton, H. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotechnol. 1996, 14, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Harbers, M.; Carninci, P. Tag-based approaches for transcriptome research and genome annotation. Nat. Methods 2005, 2, 495–502. [Google Scholar] [CrossRef]
- Haas, R.; Zelezniak, A.; Lacovacci, J.; Kamrad, S.; Townsend, S.; Ralser, M. Designing and interpreting “multi-omic” experiments that may change our understanding of biology. Curr. Opin. Syst. Biol. 2017, 6, 37–45. [Google Scholar] [CrossRef]
- Han, Y.; Gao, S.; Muegge, K.; Zhang, W.; Zhou, B. Advanced applications of RNA sequencing and challenges. Bioinform. Biol. Insights 2015, 9, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Bai, M.; Xiang, L.; Zhang, G.; Ma, W.; Jiang, H. Comparative transcriptome profiling of longissimus muscle tissues from Qianhua mutton merino and small tail Han sheep. Sci. Rep. 2016, 6, 33586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Xia, H.L.; Jiang, H.R.; Mao, Y.J.; Qu, K.X.; Huang, B.Z.; Gong, Y.C.; Yang, Z.P. Longissimus dorsi muscle transcriptomic analysis of Yunling and Chinese simmental cattle differing in intramuscular fat content and fatty acid composition. Genome 2018, 61, 549–558. [Google Scholar] [CrossRef]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. MRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Yao, Z.Z.; Velthoven, C.T.J.; Kunst, M.; Zhang, M.; McMillen, D.; Lee, C.; Jung, W.; Goldy, J.; Abdelhak, A.; Aitken, M.; et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature 2023, 624, 317–332. [Google Scholar] [CrossRef]
- Hickey, J.W.; Becker, W.R.; Nevins, S.A.; Horning, A.; Perez, A.E.; Zhu, C.; Zhu, B.; Wei, B.; Chiu, R.; Chen, D.C. Organization of the human intestine at single-cell resolution. Nature 2023, 619, 572–584. [Google Scholar] [CrossRef]
- Wu, J.J.; Zhu, S.; Gu, F.; Valencak, T.G.; Liu, J.X.; Sun, H.Z. Cross-tissue single-cell transcriptomic landscape reveals the key cell subtypes and their potential roles in the nutrient absorption and metabolism in dairy cattle. J. Adv. Res. 2021, 37, 1–18. [Google Scholar] [CrossRef]
- Wilkins, M. Proteomics data mining. Expert Rev. Proteom. 2009, 6, 599–603. [Google Scholar] [CrossRef]
- Beynon, R.J. The dynamics of the proteome: Strategies for measuring protein turnover on a proteome-wide scale. Brief Funct. Genom. Proteom. 2005, 3, 382–390. [Google Scholar] [CrossRef]
- Harper, J.W.; Bennett, E.J. Proteome complexity and the forces that drive proteome imbalance. Nature 2016, 537, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Timp, G. Beyond mass spectrometry, the next step in proteomics. Sci. Adv. 2020, 6, eaax8978. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. “Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Begou, O.; Gika, H.G.; Wilson, I.D.; Theodoridis, G. Hyphenated MS-based targeted approaches in metabolomics. Analyst 2017, 142, 3079–3100. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Koal, T.; Wang, Y.; Kohl, M.; Enot, D.P.; Deigner, H.P. Targeted metabolomics for biomarker discovery. Angew. Chem. Int. Ed. 2010, 49, 5426–5445. [Google Scholar] [CrossRef] [PubMed]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; Mclean, J.A. Untargeted metabolomics strategies—Challenges and emerging directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef]
- Hu, T.; Zhang, J.L. Mass-spectrometry-based lipidomics. J. Sep. Sci. 2018, 41, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Thaysen-Andersen, M.; Kolarich, D.; Packer, N. Glycomics & glycoproteomics: From analytics to function. Mol. Omics. 2021, 17, 8–10. [Google Scholar] [CrossRef]
- Petras, D.; Jarmusch, A.K.; Dorrestein, P.C. From single cells to our planet—Recent advances in using mass spectrometry for spatially resolved metabolomics. Curr. Opin. Chem. Biol. 2017, 36, 24–31. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef]
- Evans, H.C.; Nicodemus, M.C.; Hitit, M.; Ugur, M.R.; Kaya, A.; Topper, E.; Nicodemus, M.C.; Smith, G.D.; Memili, E. Lipidomic markers of sperm cryotolerance in cattle. Sci. Rep. 2020, 10, 20192. [Google Scholar] [CrossRef]
- O’Neil, E.V.; Spencer, T.E. Insights into the lipidome and primary metabolome of the uterus from day 14 cyclic and pregnant sheep. Biol. Reprod. 2021, 105, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Goldansaz, S.A.; Markus, S.; Plastow, G.; Wishart, D.S. Predictive blood biomarkers of sheep pregnancy and litter size. Sci. Rep. 2022, 12, 10307. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Arevalo, P.; Vaninsberghe, D.; Elsherbini, J.; Gore, J.; Polz, M.F. A reverse ecology approach based on a biological definition of microbial populations. Cell 2019, 178, 820–834.e14. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Kim, J.; Sadowsky, M.J.; Unno, T. Microbial source tracking using metagenomics and other new technologies. J. Microbiol. 2021, 59, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Shakya, A.; Lo, C.C.; Chain, P.S.G. Advances and challenges in metatranscriptomic analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.A.; Tong, Z.; Chen, R.; Pan, S. Metaproteomics study of the gut microbiome: Methods and protocols. Methods Mol. Biol. 2019, 1871, 123–132. [Google Scholar] [CrossRef]
- Hungate, R.E. The Rumen and Its Microbes; Elsevier: Amsterdam, The Netherlands, 1966; pp. 466–525. [Google Scholar]
- He, B.; Jin, S.; Cao, J.; Mi, L.; Wang, J. Metatranscriptomics of the hu sheep rumen microbiome reveals novel cellulases. Biotechnol. Biofuels 2019, 12, 153. [Google Scholar] [CrossRef]
- Xie, F.; Xu, L.; Wang, Y.; Mao, S. Metagenomic sequencing reveals that high-grain feeding alters the composition and metabolism of cecal microbiota and induces cecal mucosal injury in sheep. mSystems 2021, 6, e0091521. [Google Scholar] [CrossRef]
- Jiang, Q.; Lin, L.; Xie, F.; Jin, W.; Zhu, W.; Wang, M.; Qiu, Q.; Li, Z.; Liu, J.; Mao, S. Metagenomic insights into the microbe-mediated b and k2 vitamin biosynthesis in the gastrointestinal microbiome of ruminants. Microbiome 2022, 10, 109. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Xiong, Z.B.; Li, M.; Zheng, N.; Zhao, S.G.; Wang, J.Q. Activity- and enrichment-based metaproteomics insights into active urease from the rumen microbiota of cattle. Int. J. Mol. Sci. 2022, 23, 817. [Google Scholar] [CrossRef] [PubMed]
- Wanichthanarak, K.; Fahrmann, J.F.; Grapov, D. Genomic, proteomic, and metabolomic data integration strategies. Biomark. Insights 2015, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Ko, E.; Mersha, T.B. A roadmap for multi-omics data integration using deep learning. Brief Bioinform. 2022, 23, bbab454. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Zhu, X.W.; Mo, X.B.; Wu, L.F.; Wu, J.; Guo, Y.F.; Zeng, K.Q.; Wang, M.J.; Lin, X.; Qiu, Y.H.; et al. Integrative multi-omics analysis revealed Snp-lncRNA-mRNA (SLM) networks in human peripheral blood mononuclear cells. Hum. Genet. 2017, 136, 451–462. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Shen, M.; Xie, X.L.; Liu, G.J.; Xu, Y.X.; Lv, F.H.; Yang, H.; Yang, Y.L.; Liu, C.B.; et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef]
- Pietzner, M.; Wheeler, E.; Wheeler, E.; Cortes, A.; Koprulu, M.; Wörheide, M.A.; Oerton, E.; Cook, J.; Stewart, I.D.; Kerrison, N.D.; et al. Mapping the proteo-genomic convergence of human diseases. Science 2021, 374, eabj1541. [Google Scholar] [CrossRef]
- Noack, F.; Vangelisti, S.; Ditzer, N.; Chong, F.; Albert, M.; Bonev, B. Joint epigenome profiling reveals cell-type-specific gene regulatory programmes in human cortical organoids. Nat. Cell. Biol. 2023, 25, 1873–1883. [Google Scholar] [CrossRef]
- Perera, B.P.U.; Faulk, C.; Svoboda, L.K.; Goodrich, J.M.; Dolinoy, D.C. The role of environmental exposures and the epigenome in health and disease. Environ. Mol. Mutagen. 2020, 61, 176–192. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, X.; Zhang, Y.; Zhang, H.; Zhang, H. Comparative transcriptomic and proteomic analyses provide insights into functional genes for hypoxic adaptation in embryos of tibetan chickens. Sci. Rep. 2020, 10, 11213. [Google Scholar] [CrossRef]
- Zhao, J.Y.; Zhao, X.T.; Sun, J.T.; Zou, L.F.; Yang, S.X.; Han, X.; Zhu, W.C.; Yin, Q.; Hong, X.Y. Transcriptome and proteome analyses reveal complex mechanisms of reproductive diapause in the two-spotted spider mite, tetranychus urticae. Insect. Mol. Biol. 2017, 26, 215–232. [Google Scholar] [CrossRef]
- Xie, Y.C.; Sahin, M.; Sinha, S.; Wang, Y.F.; Nargund, A.M.; Lyu, Y.; Han, S.; Dong, Y.Y.; Hsieh, J.J.; Leslie, C.S. SETD2 loss perturbs the kidney cancer epigenetic landscape to promote metastasis and engenders actionable dependencies on histone chaperone complexes. Nat. Cancer 2022, 3, 188–202. [Google Scholar] [CrossRef]
- Serag, A.; Shakkour, Z.; Halboup, A.; Kobeissy, F.; Farag, M. Sweat metabolome and proteome: Recent trends in analytical advances and potential biological functions. J. Proteom. 2021, 246, 104310. [Google Scholar] [CrossRef]
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H.; et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese nafld. Nat. Commun. 2020, 11, 4982. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yue, S.; Yang, Z.; Feng, W.; Meng, X.; Wang, A.; Peng, C.; Wang, C.Y.; Yan, D. Oral hydroxysafflor yellow a reduces obesity in mice by modulating the gut microbiota and serum metabolism. Pharmacol. Res. 2018, 134, 40–50. [Google Scholar] [CrossRef]
- Torres, A.; Goloboff, P.A.; Catalano, S.A. Assessing topological congruence among concatenation-based phylogenomic approaches in empirical datasets. Mol. Phylogenet. Evol. 2021, 161, 107086. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, M.; Deprez, M.; Balelli, I.; Aguila, A.L.; Altmann, A. Integration of Multimodal Data. In Machine Learning for Brain Disorders; Humana: New York, NY, USA, 2023; pp. 573–597. [Google Scholar]
- Kim, Y.; Park, J.H.; Cho, Y.R. Network-based approaches for disease-gene association prediction using protein-protein interaction networks. Int. J. Mol. Sci. 2022, 23, 7411. [Google Scholar] [CrossRef]
- Qi, G.; Huang, S.; Lai, D.; Li, J.; Zhao, Y.; Shen, C.; Huang, J.; Liu, T.; Wei, K.; Wei, K.; et al. An improved joint non-negative matrix factorization for identifying surgical treatment timing of neonatal necrotizing enterocolitis. Bosn. J. Basic Med. Sci. 2022, 22, 972–981. [Google Scholar] [CrossRef]
- Wang, T.; You, J.; Gong, X.; Yang, S.; Wang, L.; Chang, Z. Probabilistic bayesian deep learning approach for online forecasting of fed-batch fermentation. ACS Omega 2023, 8, 25272–25278. [Google Scholar] [CrossRef] [PubMed]
- El-Khatib, H.; Popescu, D.; Ichim, L. Deep learning–based methods for automatic diagnosis of skin lesions. Sensor 2020, 20, 1753. [Google Scholar] [CrossRef]
- Chalise, P.; Kwon, D.; Fridley, B.; Mo, Q. Statistical methods for integrative clustering of multi-omics data. Methods Mol. Biol. 2023, 2629, 73–93. [Google Scholar] [CrossRef]
- Hossein-Zadeh, N.G. An overview of recent technological developments in bovine genomics. Vet. Anim. Sci. 2024, 25, 100382. [Google Scholar] [CrossRef] [PubMed]
- Lagoumintzis, G.; Patrinos, G.P. Triangulating nutrigenomics, metabolomics and microbiomics toward personalized nutrition and healthy living. Hum. Genom. 2023, 17, 109. [Google Scholar] [CrossRef]
- Safarlou, C.W.; Jongsma, K.R.; Vermeulen, R.; Bredenoord, A.L. The ethical aspects of exposome research: A systematic review. Exposome 2023, 3, osad004. [Google Scholar] [CrossRef]
- Wang, S.; Raza, S.H.A.; Zhang, K.; Mei, C.; Alamoudi, M.O.; Aloufi, B.H.; Alshammari, A.M.; Zan, L. Selection signatures of qinchuan cattle based on whole-genome sequences. Anim. Biotechnol. 2023, 34, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Doherty, R.; Farrelly, C.O.; Meade, K.G. Comparative epigenetics: Relevance to the regulation of production and health traits in cattle. Anim. Genet. 2014, 45, 3–14. [Google Scholar] [CrossRef]
- Fan, Y.; Liang, Y.; Deng, K.; Zhang, Z.; Wang, F. Analysis of DNA methylation profiles during sheep skeletal muscle development using whole-genome bisulfite sequencing. BMC Genom. 2020, 21, 327. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Anderson, D.F.; Brace, R.A. Multiomics analyses of vesicular transport pathway-specific transcripts and proteins in ovine amnion: Responses to altered intramembranous transport. Physiol. Genom. 2019, 51, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Raza, S.H.A.; Du, X.; Wang, J.; Wang, M.; Ma, J.; Xie, K.; Pant, S.D.; He, J.; Aloufi, B.H.; et al. Effect of feeding corn silage on semen quality and spermatogenesis of bulls. Vet. Res. Commun. 2024, 48, 391–401. [Google Scholar] [CrossRef]
- Wu, G.; Qiu, X.; Jiao, Z.; Yang, W.; Pan, H.; Li, H.; Bian, Z.; Geng, Q.; Wu, H.; Jiang, J.; et al. Integrated analysis of transcriptome and metabolome profiles in the longissimus dorsi muscle of buffalo and cattle. Curr. Issues Mol. Biol. 2023, 45, 9723–9736. [Google Scholar] [CrossRef]
- Ma, D.; Yu, Q.; Hedrick, V.E.; Cooper, B.R.; Sobreira, P.; Oh, J.H.; Chun, H.; Kim, Y.H.B. Proteomic and metabolomic profiling reveals the involvement of apoptosis in meat quality characteristics of ovine m. longissimus from different callipyge genotypes. Meat Sci. 2020, 166, 108140. [Google Scholar] [CrossRef]
- Zhao, B.; Luo, H.; He, J.; Huang, X.; Chen, S.; Fu, X.; Zeng, W.; Tian, Y.; Liu, S.; Li, C.J. Comprehensive transcriptome and methylome analysis delineates the biological basis of hair follicle development and wool-related traits in merino sheep. BMC Biol. 2021, 19, 197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, F.; Zhang, X.; Zhang, D.; Li, X.; Zhang, Y.; Zhao, Y.; Song, Q.; Huang, K.; Xu, D.; et al. Integrative analysis of transcriptomics and proteomics of longissimus thoracis of the hu sheep compared with the dorper sheep. Meat Sci. 2022, 193, 108930. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Wang, N.; Li, Z.; Heller, R.; Liu, R.; Zhao, Y.; Han, J.; Pan, X.; Zheng, Z.; et al. Genetic basis of ruminant headgear and rapid antler regeneration. Science 2019, 364, eaav6335. [Google Scholar] [CrossRef]
- Wu, J.; Yang, D.; Gong, H.; Qi, Y.; Qiu, X. Multiple omics analysis reveals that high fiber diets promote gluconeogenesis and inhibit glycolysis in muscle. BMC Genom. 2020, 21, 660. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, X.; Li, X.; Zheng, J.; Zhao, L.; Fan, C.; Zhao, Y. Integrated metabolomics and transcriptomics analyses reveal the candidate genes regulating the meat quality change by castration in Yudong black goats (Capra hircus). Genes 2023, 15, 43. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, H.; Li, L.; Tan, J.; Wang, Y.; Liu, M.; Jiang, L. Multi-omics analysis reveals that the metabolite profile of raw milk is associated with dairy cows’ health status. Food Chem. 2023, 428, 136813. [Google Scholar] [CrossRef]
- Deng, J.; Liu, Y.J.; Wei, W.T.; Huang, Q.X.; Zhao, L.P.; Luo, L.Y.; Zhu, Q.; Zhang, L.; Chen, Y.; Ren, Y.L.; et al. Single-cell transcriptome and metagenome profiling reveals the genetic basis of rumen functions and convergent developmental patterns in ruminants. Genome Res. 2023, 33, 1690–1707. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Han, L.; Zhang, S.; Zhang, X.; Hou, S.; Gui, L.; Sun, S.; Yuan, Z.; Wang, Z.; Wang, Z. Insight into the differences of meat quality between Qinghai white Tibetan sheep and black Tibetan sheep from the perspective of metabolomics and rumen microbiota. Food Chem. X 2023, 19, 100843. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, L.; Jiang, X.; Song, Y.; Wang, D.; Liu, H.; Wu, S.; Yao, J. Multiomics analysis revealed that the metabolite profile of raw milk is associated with lactation stage of dairy cows and could be affected by variations in the ruminal microbiota. J. Dairy Sci. 2024. in Press. [Google Scholar] [CrossRef]
- Li, S.; Mu, R.; Zhu, Y.; Zhao, F.; Qiu, Q.; Si, H.; Wright, A.-D.G.; Li, Z. Shifts in the microbial community and metabolome in rumen ecological niches during antler growth. Comput. Struct. Biotechnol. J 2024, 23, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Whon, T.W.; Sung, H.; Jeong, Y.S.; Jung, E.S.; Shin, N.R.; Hyun, D.W.; Kim, P.S.; Lee, J.Y.; Lee, C.H. Longitudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat. Commun. 2021, 12, 161. [Google Scholar] [CrossRef]
- Eloiflin, R.J.; Auray, G.; Python, S.; Rodrigues, V.; Seveno, M.; Urbach, S.; El Koulali, K.; Holzmuller, P.; Totte, P.; Libeau, G. Identification of Differential Responses of Goat PBMCs to PPRV Virulence Using a Multi-Omics Approach. Front. Immunol. 2021, 12, 745315. [Google Scholar] [CrossRef]
- Hall, T.J.; McHugo, G.P.; Mullen, M.P.; Ward, J.A.; Killick, K.E.; Browne, J.A.; Gordon, S.V.; MacHugh, D.E. Integrative and comparative genomic analyses of mammalian macrophage responses to intracellular mycobacterial pathogens. Tuberculosis 2024, 147, 102453. [Google Scholar] [CrossRef] [PubMed]
- Naserkheil, M.; Ghafouri, F.; Zakizadeh, S.; Pirany, N.; Zeinab, M.; Ghorbani, S.; Banabazi, M.H.; Bakhtiarizadeh, M.R.; Huq, M.A.; Park, M.N. Multi-omics integration and network analysis reveal potential hub genes and genetic mechanisms regulating bovine mastitis. Curr. Issues Mol. Biol. 2022, 44, 309–328. [Google Scholar] [CrossRef]
- Xu, Y.X.; Wang, B.; Jing, J.N.; Ma, R.; Luo, Y.; Li, X.; Yan, Z.; Liu, Y.J.; Gao, L.; Ren, Y.L. Whole-body adipose tissue multi-omic analyses in sheep reveal molecular mechanisms underlying local adaptation to extreme environments. Commun. Biol. 2023, 6, 159. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, Y.; Yang, J.; Dong, L.; Zhang, R.; Tian, S.; Yu, Y.; Ren, L.; Hou, W.; Zhu, F.; et al. Multi-omics data integration using ratio-based quantitative profiling with Quartet reference materials. Nat. Biotechnol. 2024, 42, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Chafai, N.; Hayah, I.; Houaga, I.; Badaoui, B. A review of machine learning models applied to genomic prediction in animal breeding. Front. Genet. 2023, 14, 1150596. [Google Scholar] [CrossRef]
- O’Hara, E.; Neves, A.L.; Song, Y.; Guan, L.L. The role of the gut microbiome in cattle production and health: Driver or passenger? Annu. Rev. Anim. Biosci. 2020, 8, 199–220. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Omics | Pan-Omics Integrative Analysis | Function | References |
|---|---|---|---|
| Genomics | Transcriptomics | (1) Gene expression regulation: Effects of gene structural variations (SNP, CNV, SV, etc.) on gene expression. (2) Identification of functional variants: Finding gene variants that significantly impact the organism’s function. (3) Developmental and tissue-specific insights: Revealing gene expression patterns in different tissues or developmental stages. | [87,88] |
| Proteomics | (1) Correlation between genotype and phenotype and functional validation: Analyzing the relationship between gene variants and protein expression levels and post-translational modifications, and verifying the functional changes at the protein level caused by sequence variations. (2) Complex trait analysis and biomarker discovery: Dissecting the genetic variations in traits caused by changes in protein networks, and identifying protein biomarkers influenced by gene variants. | [89] | |
| Epigenomics Transcriptomics | (1) Regulation of gene activity and phenotypic variation: Elucidating the effects of DNA methylation, histone modifications, and chromatin accessibility on gene expression and genome stability to obtain different phenotypes. (2) Environmental interactions: Exploring the regulation of gene expression and phenotypic changes by environmental factors. | [90,91] | |
| Transcriptomics | Proteomics | (1) Correlation between mRNA and protein levels and functional validation: Understanding the mechanisms and effects of post-transcriptional regulation of mRNA, and verifying the relationship between gene expression and protein levels and activity. (2) Complex trait analysis: Gaining a more comprehensive understanding of functional protein changes in the regulatory mechanisms of gene expression changes. | [92,93] |
| Metagenomics Metabolomics | (1) Regulation of metabolic products by the combined effects of gene expression and the microbiome. (2) Revealing the roles and impacts of microorganisms in host metabolic processes. (3) Discovering the interactions between gene expression processes, the microbiome, and metabolites on the physiological state of the organism. (4) Providing a deeper understanding of the internal regulatory networks and their complexity within the organism. | [93,94] | |
| Epigenomics | (1) Gene expression regulation: Analyzing how epigenetic modifications (DNA methylation, histone modifications, etc.) regulate gene expression. (2) Understanding cellular differentiation and different physiological mechanisms: Revealing the processes of cell and tissue development and identifying epigenetic dysregulation associated with abnormal gene expression in various physiological states. | [94] | |
| Metabolomics | Proteomics | Analyzing the molecular mechanisms and regulatory pathways of different omics in response to external stimuli, to more intuitively understand the upstream and downstream regulatory relationships between metabolites, enzymes, and genes. | [95] |
| Metagenomics | (1) Identifying metabolites produced by the microbiome and their impact on host metabolic pathways. (2) Elucidating the relationship between microbial diversity and function and host metabolic health. | [96,97] |
| Data Integration Methodology | Advantages | Disadvantages | Typical Methods | References |
|---|---|---|---|---|
| Connection-based methods | Simple to apply and does not require complex transformations | Increased computational complexity; unable to effectively capture unique structures and relationships of data types | Multi-Omics Factor Analysis (MOFA); iCluster | [98] |
| Transformation-based methods | Effectively identifies and captures correlations between datasets; simplifies analysis through low-dimensional space | Unable to capture complex biological interactions; the transformation process is computationally demanding | Canonical Correlation Analysis (CCA); Partial Least Squares (PLS) | [99] |
| Network construction-based methods | Effectively captures interactions and relationships between datasets; visually interprets biological data intuitively | Building and integrating multiple networks is computationally and conceptually complex; limited scalability | Similarity Network Fusion (SNF); Multi-Omics Graph Convolutional Network (MOGCN) | [100] |
| Matrix factorization-based methods | Effectively reduces dimensions while capturing underlying patterns and structures; can handle various types of omics data and missing values | The decomposed components may be difficult to interpret biologically | Joint Non-Negative Matrix Factorization (NMF); Multi-Omics Tensor Decomposition | [101] |
| Bayesian and probabilistic methods | Effectively models uncertainty and variability in data; can capture complex, non-linear relationships | Computationally demanding; requires expertise in probabilistic modeling and Bayesian statistics | Bayesian Network Models; Multi-Omics Factor Analysis (MOFA+) | [102] |
| Deep learning-based methods | Can capture complex, non-linear relationships between omics datasets; automatically learns relevant features and representations from data | High data requirements; deep learning models are challenging to interpret biologically | Deep Learning Autoencoders; Variational Autoencoders (VAE) | [103] |
| Hybrid methods | Leverages multiple integration strategies to improve overall performance | Increased complexity; balancing and integrating results from different methods is challenging | Integrative Clustering (iClusterPlus); Multi-Omics Factor Analysis via Transfer Learning | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Z.; Lu, Y.; Li, M.; Chong, Y.; Hong, J.; Wu, J.; Wu, D.; Xi, D.; Deng, W. Application of Pan-Omics Technologies in Research on Important Economic Traits for Ruminants. Int. J. Mol. Sci. 2024, 25, 9271. https://doi.org/10.3390/ijms25179271
Gao Z, Lu Y, Li M, Chong Y, Hong J, Wu J, Wu D, Xi D, Deng W. Application of Pan-Omics Technologies in Research on Important Economic Traits for Ruminants. International Journal of Molecular Sciences. 2024; 25(17):9271. https://doi.org/10.3390/ijms25179271
Chicago/Turabian StyleGao, Zhendong, Ying Lu, Mengfei Li, Yuqing Chong, Jieyun Hong, Jiao Wu, Dongwang Wu, Dongmei Xi, and Weidong Deng. 2024. "Application of Pan-Omics Technologies in Research on Important Economic Traits for Ruminants" International Journal of Molecular Sciences 25, no. 17: 9271. https://doi.org/10.3390/ijms25179271
APA StyleGao, Z., Lu, Y., Li, M., Chong, Y., Hong, J., Wu, J., Wu, D., Xi, D., & Deng, W. (2024). Application of Pan-Omics Technologies in Research on Important Economic Traits for Ruminants. International Journal of Molecular Sciences, 25(17), 9271. https://doi.org/10.3390/ijms25179271