
A Cautionary Tale of Hypertrophic Cardiomyopathy—From “Benign” Left Ventricular Hypertrophy to Stroke, Atrial Fibrillation, and Molecular Genetic Diagnostics: A Case Report and Review of Literature
 , ,
, ,
Abstract
:1. Introduction
2. Detailed Case Description
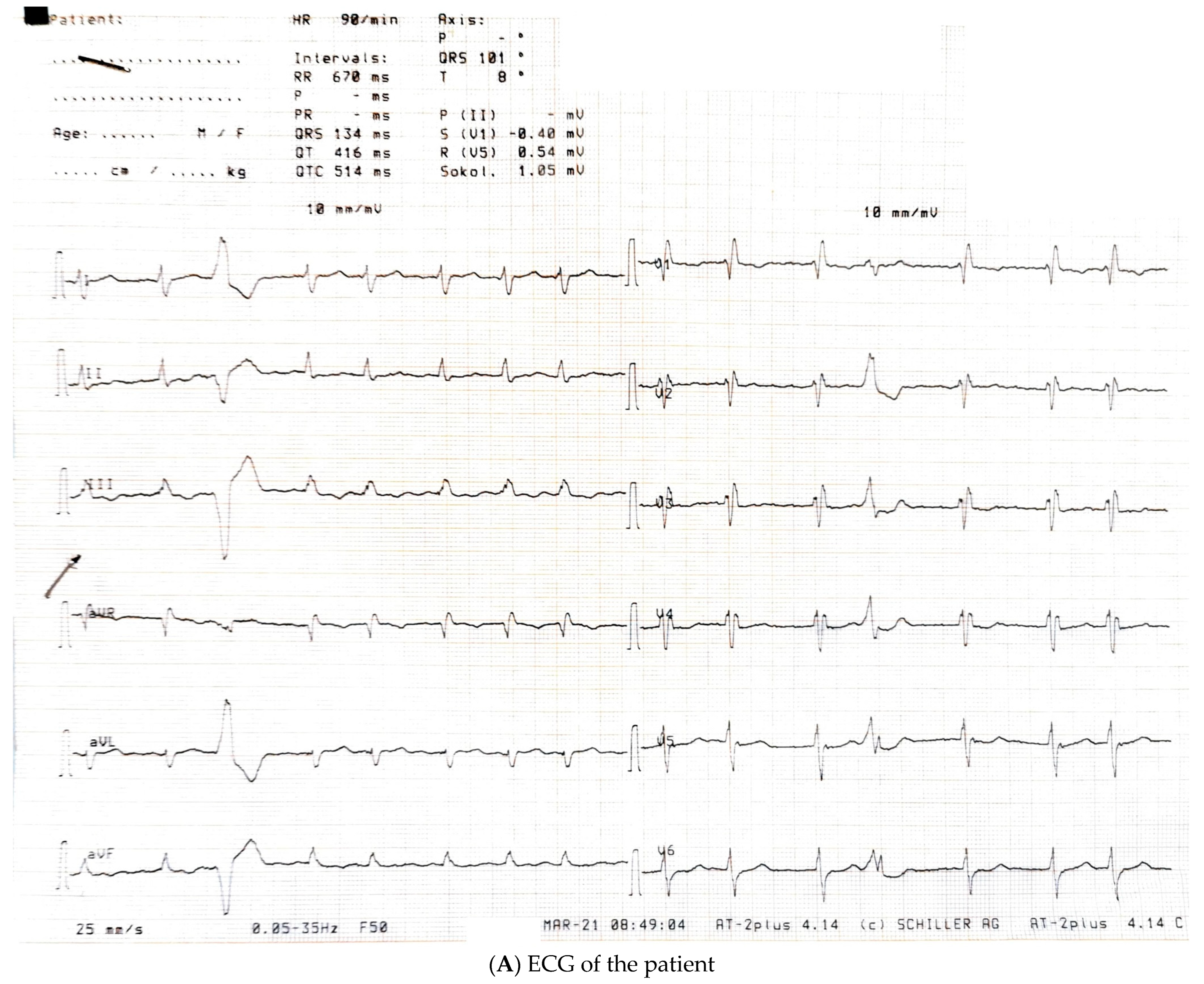
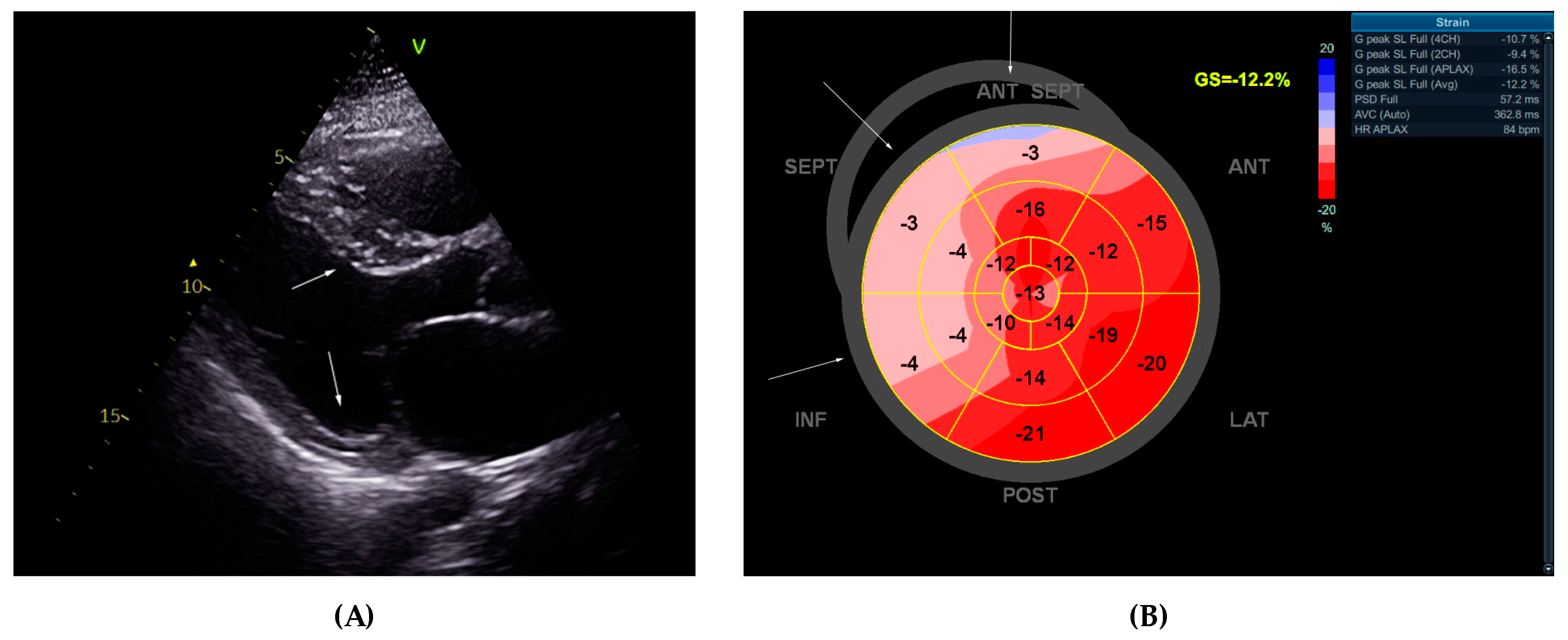
2.1. Echocardiographic Study
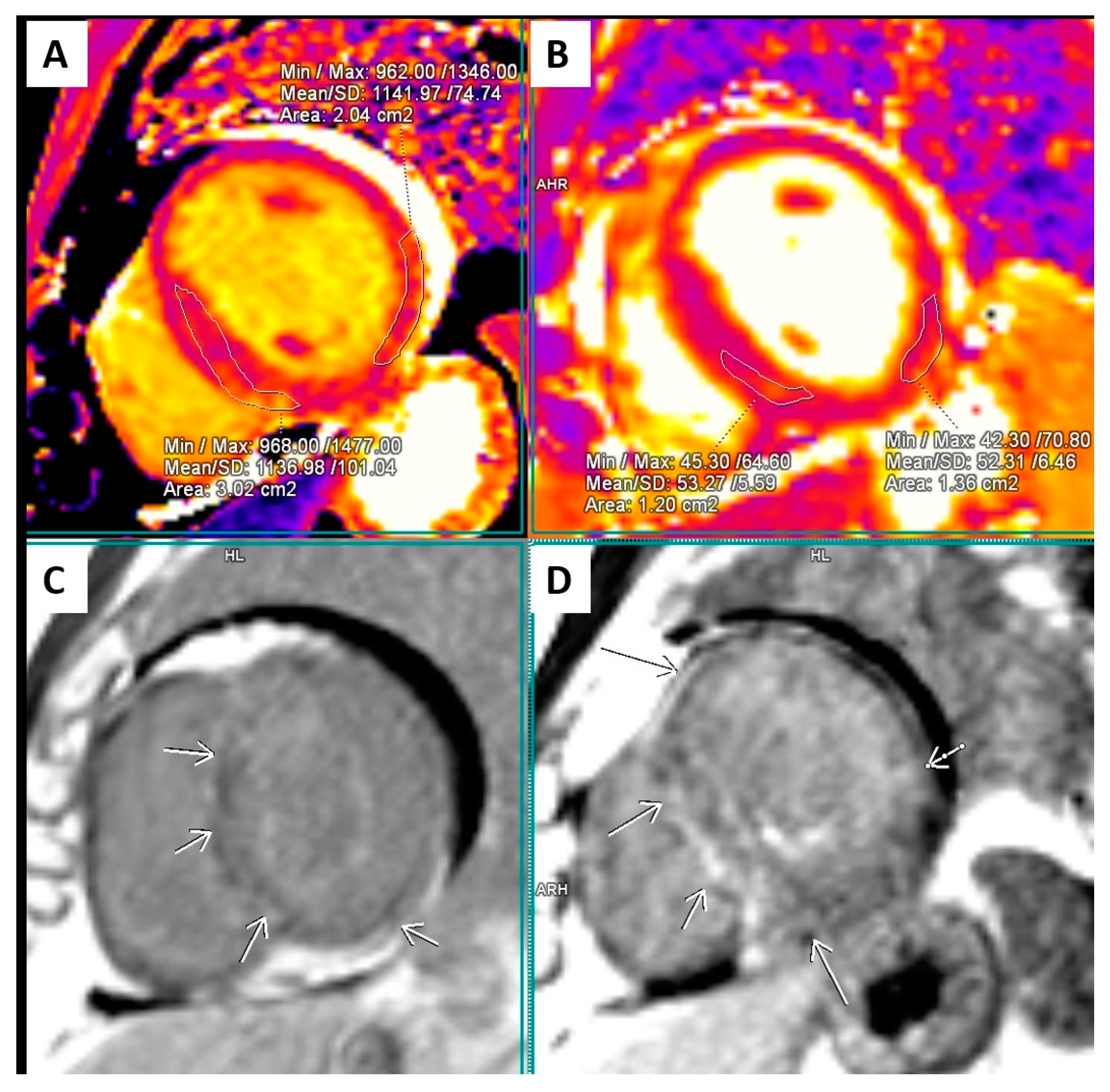
2.2. Cardiac Magnetic Resonance (CMR)
2.3. Follow-Up
2.4. Molecular-Genetic Analysis
2.5. Diagnosis and Further Follow-Up
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef]
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. In Genetic Causes of Cardiac Disease. Cardiac and Vascular Biology; Erdmann, J., Moretti, A., Eds.; Springer: Cham, Switzerland, 2019; Volume 7. [Google Scholar] [CrossRef]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ. Cardiovasc. Genet. 2017, 10, e001620. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.M.; Will, M.L.; Gersh, B.J.; Kruisselbrink, T.M.; Ommen, S.R.; Ackerman, M.J. Characterization of a phenotype-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin. Proc. 2014, 89, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Akhtar, M.; Elliott, P. The genetics of hypertrophic cardiomyopathy. Glob. Cardiol. Sci. Pract. 2018, 2018, 36. [Google Scholar] [CrossRef]
- Ingles, J.; Doolan, A.; Chiu, C.; Seidman, J.; Seidman, C.; Semsarian, C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. J. Med. Genet. 2005, 42, e59. [Google Scholar] [CrossRef]
- Kelly, M.; Semsarian, C. Multiple mutations in genetic cardiovascular disease: A marker of disease severity? Circ. Cardiovasc. Genet. 2009, 2, 182–190. [Google Scholar] [CrossRef]
- Girolami, F.; Ho, C.Y.; Semsarian, C.; Baldi, M.; Will, M.L.; Baldini, K.; Torricelli, F.; Yeates, L.; Cecchi, F.; Ackerman, M.J.; et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J. Am. Coll. Cardiol. 2010, 55, 1444–1453. [Google Scholar] [CrossRef]
- Vitale, G.; Coppini, R.; Tesi, C.; Poggesi, C.; Sacconi, L.; Ferrantini, C. T-tubule remodeling in human hypertrophic cardiomyopathy. J. Muscle Res. Cell Motil. 2021, 42, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Tejado, B.S.M.; Jou, C. Histopathology in HCM. Glob. Cardiol. Sci. Pract. 2018, 2018, 20. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. ESC Scientific Document Group, 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Ommen, S.; Ho, C.; Asif, I.M.; Balaji, S.; Burke, M.A.; Day, S.M.; Dearani, J.A.; Epps, K.C.; Evanovich, L.; Ferrari, V.A.; et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. JACC 2024, 83, 2324–2405. [Google Scholar] [CrossRef] [PubMed]
- Corona-Villalobos, C.P.; Sorensen, L.; Chu, L.; Abraham, T.; Kamel, I.R.; Zimmerman, S.L. Maximal left ventricular wall thickness: A comparison between CMR and echocardiography in hypertrophic cardiomyopathy. J. Cardiovasc. Magn. Reson. 2013, 15 (Suppl. S1), 169. [Google Scholar] [CrossRef]
- Girolami, F.; Gozzini, A.; Pálinkás, E.D.; Ballerini, A.; Tomberli, A.; Baldini, K.; Marchi, A.; Zampieri, M.; Passantino, S.; Porcedda, G.; et al. Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions. J. Clin. Med. 2023, 12, 2489. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Kiaos, A.; Daskalopoulos, G.N.; Kamperidis, V.; Ziakas, A.; Efthimiadis, G.; Karamitsos, T.D. Quantitative Late Gadolinium Enhancement Cardiac Magnetic Resonance and Sudden Death in Hypertrophic Cardiomyopathy: A Meta-Analysis. JACC Cardiovasc. Imaging 2024, 17, 489–497. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726, Erratum in Eur. Heart J. 2021, 42, 4901. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2023, 44, 3627–3639, Erratum in Eur. Heart J. 2024, 45, 53. [Google Scholar] [CrossRef]
- Cacciapuoti, F. Molecular mechanisms of left ventricular hypertrophy (LVH) in systemic hypertension (SH)—Possible therapeutic perspectives. J. Am. Soc. Hypertens. 2011, 5, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Canadilla, P.; Cook, A.C.; Mohun, T.J.; Oji, O.; Schlossarek, S.; Carrier, L.; McKenna, W.J.; Moon, J.C.; Captur, G. Myoarchitectural disarray of hypertrophic cardiomyopathy begins pre-birth. J. Anat. 2019, 235, 962–976. [Google Scholar] [CrossRef]
- Tripathi, B.; Khan, S.; Arora, S.; Kumar, V.; Naraparaju, V.; Lahewala, S.; Sharma, P.; Atti, V.; Jain, V.; Shah, M.; et al. Burden and trends of arrhythmias in hypertrophic cardiomyopathy and its impact of mortality and resource utilization. J. Arrhythm. 2019, 35, 612–625. [Google Scholar] [CrossRef]
- Abdelfattah, O.M.; Martinez, M.; Sayed, A.; ElRefaei, M.; Abushouk, A.I.; Hassan, A.; Masri, A.; Winters, S.L.; Kapadia, S.R.; Maron, B.J.; et al. Temporal and Global Trends of the Incidence of Sudden Cardiac Death in Hypertrophic Cardiomyopathy. JACC Clin. Electrophysiol. 2022, 8, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Stroumpoulis, K.I.; Pantazopoulos, I.N.; Xanthos, T.T. Hypertrophic cardiomyopathy and sudden cardiac death. World J. Cardiol. 2010, 2, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Mistrulli, R.; Ferrera, A.; Muthukkattil, M.L.; Battistoni, A.; Gallo, G.; Barbato, E.; Spera, F.R.; Magrì, D. Atrial Fibrillation in Patients with Hypertrophic Cardiomyopathy and Cardiac Amyloidosis: From Clinical Management to Catheter Ablation Indication. J. Clin. Med. 2024, 13, 501. [Google Scholar] [CrossRef]
- Penela, D.; Sorgente, A.; Cappato, R. State-of-the-Art Treatments for Atrial Fibrillation in Patients with Hypertrophic Cardiomyopathy. J. Clin. Med. 2021, 10, 3025. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar]
- Guttmann, O.P.; Rahman, M.S.; O’Mahony, C.; Anastasakis, A.; Elliott, P.M. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: Systematic review. Heart 2014, 100, 465–472. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Z.; Chen, X.; He, S. Thromboembolism in Patients with Hypertrophic Cardiomyopathy. Int. J. Med. Sci. 2021, 18, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, C.; Zocchi, C.; Ciabatti, M.; Milazzo, A.; Cappelli, F.; Fumagalli, S.; Pieroni, M.; Olivotto, I. From Atrial Fibrillation Management to Atrial Myopathy Assessment: The Evolving Concept of Left Atrium Disease in Hypertrophic Cardiomyopathy. Can. J. Cardiol. 2024, 40, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Pioner, J.M.; Vitale, G.; Gentile, F.; Scellini, B.; Piroddi, N.; Cerbai, E.; Olivotto, I.; Tardiff, J.; Coppini, R.; Tesi, C.; et al. Genotype-Driven Pathogenesis of Atrial Fibrillation in Hypertrophic Cardiomyopathy: The Case of Different TNNT2 Mutations. Front. Physiol. 2022, 13, 864547. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.F.; Gandhi, S.; Siegel, R.J.; Rader, F. Anticoagulation for stroke prevention in patients with hypertrophic cardiomyopathy and atrial fibrillation: A review. Heart Rhythm. 2021, 18, 297–302. [Google Scholar] [CrossRef]
- Marszalek, R.J.; Solaro, R.J.; Wolska, B.M. Coronary arterial vasculature in the pathophysiology of hypertrophic cardiomyopathy. Pflug. Arch. 2019, 471, 769–780. [Google Scholar] [CrossRef]
- Foà, A.; Agostini, V.; Rapezzi, C.; Olivotto, I.; Corti, B.; Potena, L.; Biagini, E.; Suarez, S.M.; Rotellini, M.; Cecchi, F.; et al. Histopathological comparison of intramural coronary artery remodeling and myocardial fibrosis in obstructive versus end-stage hypertrophic cardiomyopathy. Int. J. Cardiol. 2019, 291, 77–82. [Google Scholar] [CrossRef]
- Achim, A.; Savaria, B.U.; Buja, L.M. Commentary on the enigma of small vessel disease in hypertrophic cardiomyopathy: Is invasive assessment of microvascular resistance a novel independent predictor of prognosis? Cardiovasc. Pathol. 2022, 60, 107448. [Google Scholar] [CrossRef] [PubMed]
- Aksakal, E.; Yapici, O.; Yazici, M.; Yilmaz, O.; Sahin, M. Apical hypertrophic cardiomyopathy: A case of slow flow in lad and malign ventricular arrhythmia. Int. J. Cardiovasc. Imaging 2005, 21, 185–188. [Google Scholar] [CrossRef]
- Mood, E.P.; Kahnooji, M.; Aliramezany, M. Ectasia and slow flow phenomena of coronary artery related to apical hypertrophic cardiomyopathy. Clin. Case Rep. 2023, 11, e7870. [Google Scholar] [CrossRef]
- Candemir, M.; Şahinarslan, A.; Yazol, M.; Öner, Y.A.; Boyacı, B. Determination of Myocardial Scar Tissue in Coronary Slow Flow Phenomenon and the Relationship Between Amount of Scar Tissue and Nt-ProBNP. Determinação do Tecido Cicatricial do Miocárdio no Fenômeno de Fluxo Coronário Lento e a Relação entre a Quantidade de Tecido Cicatricial e o Nt-ProBNP. Arq. Bras. Cardiol. 2020, 114, 540–551. [Google Scholar] [CrossRef]
- Bernardini, A.; Crotti, L.; Olivotto, I.; Cecchi, F. Diagnostic and prognostic electrocardiographic features in patients with hypertrophic cardiomyopathy. Eur. Heart J. Suppl. 2023, 25 (Suppl. C), C173–C178, Erratum in Eur. Heart J. Suppl. 2023, 25 (Suppl. C), suad121. Erratum in Eur. Heart J. Suppl. 2023, 25 (Suppl. C), C178. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rodrigues, J.C.L.; McIntyre, B.; Dastidar, A.G.; Lyen, S.M.; E Ratcliffe, L.; E Burchell, A.; Hart, E.C.; Bucciarelli-Ducci, C.; Hamilton, M.C.K.; Paton, J.F.R.; et al. The effect of obesity on electrocardiographic detection of hypertensive left ventricular hypertrophy: Recalibration against cardiac magnetic resonance. J. Hum. Hypertens. 2016, 30, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Bazoukis, G.; Garcia-Zamora, S.; Çinier, G.; Lee, S.; Gul, E.E.; Álvarez-García, J.; Miana, G.; Hayıroğlu, M.; Tse, G.; Liu, T.; et al. Association of electrocardiographic markers with myocardial fibrosis as assessed by cardiac magnetic resonance in different clinical settings. World J. Cardiol. 2022, 14, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.; Bueno-Orovio, A.; Zacur, E.; Ariga, R.; Grau, V.; Neubauer, S.; Watkins, H.; Rodriguez, B.; Mincholé, A. Electrocardiogram phenotypes in hypertrophic cardiomyopathy caused by distinct mechanisms: Apico-basal repolarization gradients vs. Purkinje-myocardial coupling abnormalities. Europace 2018, 20 (Suppl. S3), iii102–iii112. [Google Scholar] [CrossRef]
- Cuddy, S.A.; Chetrit, M.; Jankowski, M.; Desai, M.; Falk, R.H.; Weiner, R.B.; Klein, A.L.; Phelan, D.; Grogan, M. Practical Points for Echocardiography in Cardiac Amyloidosis. J. Am. Soc. Echocardiogr. 2022, 35, A31–A40. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S.; Maron, B.A.; Loscalzo, J. Moving Beyond the Sarcomere to Explain Heterogeneity in Hypertrophic Cardiomyopathy: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 1978–1986. [Google Scholar] [CrossRef]
- Segura-Villalobos, F.; Hernández-Guerra, A.I.; Wanguemert-Pérez, F.; Rodríguez-Pérez, J.C.; Mendoza-Lemes, H.; Barriales-Villa, R. Hypertrophic Cardiomyopathy Without Ventricular Hypertrophy: Usefulness of Genetic and Pathological Study in Preventing Sudden Death. Rev. Esp. Cardiol. 2017, 70, 604–606. [Google Scholar] [CrossRef]
- Soler, R.; Méndez, C.; Rodríguez, E.; Barriales, R.; Ochoa, J.P.; Monserrat, L. Phenotypes of hypertrophic cardiomyopathy. An illustrative review of MRI findings. Insights Imaging 2018, 9, 1007–1020. [Google Scholar] [CrossRef]
- Olson, T.M.; Doan, T.P.; Kishimoto, N.Y.; Whitby, F.G.; Ackerman, M.J.; Fananapazir, L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic car-diomyopathy. J. Mol. Cell. Cardiol. 2000, 32, 1687–1694. [Google Scholar] [CrossRef]
- Olson, T.M.; Michels, V.V.; Thibodeau, S.N.; Tai, Y.-S.; Keating, M.T. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998, 280, 750–752. [Google Scholar] [CrossRef]
- Arad, M.; Penas-Lado, M.; Monserrat, L.; Maron, B.J.; Sherrid, M.; Ho, C.Y.; Barr, S.; Karim, A.; Olson, T.M.; Kamisago, M.; et al. Gene mutations in apical hypertrophic cardiomyopathy. Circulation 2005, 112, 2805–2811. [Google Scholar] [CrossRef]
- Kabsch, W.; Mannherz, H.G.; Suck, D.; Pai, E.F.; Holmes, K.C. Atomic structure of the actin:DNase I complex. Nature 1990, 347, 37–44. [Google Scholar] [CrossRef]
- Despond, E.A.; Dawson, J.F. Classifying Cardiac Actin Mutations Associated With Hypertrophic Cardiomyopathy. Front. Physiol. 2018, 9, 405. [Google Scholar] [CrossRef] [PubMed]
- Bookwalter, C.S.; Trybus, K.M. Functional consequences of a mutation in an expressed human alpha-cardiac actin at a site implicated in familial hypertrophic cardio-myopathy. J. Biol. Chem. 2006, 281, 16777–16784. [Google Scholar] [CrossRef]
- Debold, E.P.; Saber, W.; Cheema, Y.; Bookwalter, C.S.; Trybus, K.M.; Warshaw, D.M.; VanBuren, P. Human actin mutations associated with hypertrophic and dilated cardiomyopathies demonstrate distinct thin filament regulatory properties in vitro. J. Mol. Cell. Cardiol. 2010, 48, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Dahari, M.; Dawson, J.F. Do cardiac actin mutations lead to altered actomyosin interactions? Biochem. Cell Biol. 2015, 93, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Vikhorev, P.G.; Kashyap, M.N.; Rowlands, C.; Ferenczi, M.A.; Woledge, R.C.; MacLeod, K.; Marston, S.; Curtin, N.A. Mechanical and energetic properties of papillary muscle from ACTC E99K transgenic mouse models of hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1513–H1524. [Google Scholar] [CrossRef]
- Liu, H.; Henein, M.; Anillo, M.; Dawson, J.F. Cardiac actin changes in the actomyosin interface have different effects on myosin duty ratio. Biochem Cell Biol. 2018, 96, 26–31. [Google Scholar] [CrossRef]
- Song, W.; Dyer, E.; Stuckey, D.J.; Copeland, O.; Leung, M.-C.; Bayliss, C.; Messer, A.; Wilkinson, R.; Tremoleda, J.L.; Schneider, M.D.; et al. Molecular mechanism of the E99K mutation in cardiac actin (ACTC Gene) that causes apical hypertrophy in man and mouse. J. Biol. Chem. 2011, 286, 27582–27593. [Google Scholar] [CrossRef]
- Teng, G.Z.; Shaikh, Z.; Liu, H.; Dawson, J.F. M-class hypertrophic cardiomyopathy cardiac actin mutations increase calcium sensitivity of regulated thin filaments. Biochem. Biophys Res Commun. 2019, 519, 148–152. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Zou, Y.; Sun, K.; Wang, Z.; Ding, H.; Yuan, J.; Wei, W.; Hou, Q.; Wang, H.; et al. Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy. Eur. J. Heart Fail. 2014, 16, 950–957. [Google Scholar] [CrossRef]
- Murphy, S.L.; Anderson, J.H.; Kapplinger, J.D.; Kruisselbrink, T.M.; Gersh, B.J.; Ommen, S.R.; Ackerman, M.J.; Bos, J.M. Evaluation of the Mayo Clinic Phenotype-Based Genotype Predictor Score in Patients with Clinically Diagnosed Hypertrophic Cardiomyopathy. J. Cardiovasc. Transl. Res. 2016, 9, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Zarain-Herzberg, A.; Willard, H.F.; Tada, M.; MacLennan, D.H. Structure of the rabbit phospholamban gene, cloning of the human cDNA, and assignment of the gene to human chromosome 6. J. Biol. Chem. 1991, 266, 11669–11675. [Google Scholar] [CrossRef] [PubMed]
- Koss, K.L.; Kranias, E.G. Phospholamban: A prominent regulator of myocardial contractility. Circ. Res. 1996, 79, 1059–1063. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Pater, L.; Lynch, R.A.; Asahi, M.; Gramolini, A.O.; Fan, G.-C.; Tsiapras, D.; Hahn, H.S.; Adamopoulos, S.; et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Investig. 2003, 111, 869–876. [Google Scholar] [CrossRef]
- Kelly, E.M.; Hou, Z.; Bossuyt, J.; Bers, D.M.; Robia, S.L. Phospholamban oligomerization, quaternary structure, and sarco(endo)plasmic reticulum calcium ATPase binding measured by fluorescence resonance energy transfer in living cells. J. Biol. Chem. 2008, 283, 12202–12211. [Google Scholar] [CrossRef]
- Medeiros, A.; Biagi, D.G.; Sobreira, T.J.; de Oliveira, P.S.L.; Negrão, C.E.; Mansur, A.J.; Krieger, J.E.; Brum, P.C.; Pereira, A.C. Mutations in the human phospholamban gene in patients with heart failure. Am. Heart J. 2011, 162, 1088–1095.e1. [Google Scholar] [CrossRef]
- Sanoudou, D.; Kolokathis, F.; Arvanitis, D.; Al-Shafai, K.; Krishnamoorthy, N.; Buchan, R.J.; Walsh, R.; Tsiapras, D.; Barton, P.J.; A Cook, S.; et al. Genetic modifiers to the PLN L39X mutation in a patient with DCM and sustained ventricular tachycardia? Glob. Cardiol. Sci. Pract. 2015, 2015, 29. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Adekola, B.A.; Bos, J.M.; Ommen, S.R.; Ackerman, M.J. PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: Summary of the literature and implications for genetic testing. Am. Heart J. 2011, 161, 165–171. [Google Scholar] [CrossRef]
- Chiu, C.; Tebo, M.; Ingles, J.; Yeates, L.; Arthur, J.W.; Lind, J.M.; Semsarian, C. Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2007, 43, 337–343. [Google Scholar] [CrossRef]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888, Erratum in Genet. Med. 2015, 17, 319. [Google Scholar] [CrossRef] [PubMed]
- Mellor, G.; Laksman, Z.W.; Tadros, R.; Roberts, J.D.; Gerull, B.; Simpson, C.S.; Klein, G.J.; Champagne, J.; Talajic, M.; Gardner, M.; et al. Genetic Testing in the Evaluation of Unexplained Cardiac Arrest: From the CASPER (Cardiac Arrest Survivors With Preserved Ejection Fraction Registry). Circ. Cardiovasc. Genet. 2017, 10, e001686. [Google Scholar] [CrossRef]
- Ng, D.; Johnston, J.J.; Teer, J.K.; Singh, L.N.; Peller, L.C.; Wynter, J.S.; Lewis, K.L.; Cooper, D.N.; Stenson, P.D.; Mullikin, J.C.; et al. Interpreting secondary cardiac disease variants in an exome cohort. Circ. Cardiovasc. Genet. 2013, 6, 337–346. [Google Scholar] [CrossRef]
- Gellens, M.E.; George, A.L.; Chen, L.Q.; Chahine, M.; Horn, R.; Barchi, R.L.; Kallen, R.G. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. USA 1992, 89, 554–558. [Google Scholar] [CrossRef]
- Motoike, H.K.; Liu, H.; Glaaser, I.W.; Yang, A.S.; Tateyama, M.; Kass, R.S. The Na+ channel inactivation gate is a molecular complex: A novel role of the COOH-terminal domain. J. Gen. Physiol. 2004, 123, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: A potential link to sudden death in the absence of conventional risk factors. Heart Rhythm. 2012, 9, 57–63. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time/Period/Age | Clinical Presentation | Management | Therapy |
|---|---|---|---|
| Early 20s | “Hypertrophy of the heart” noted during obligatory military service | No recommendations given. Explained with increased physical activity during military service. | |
| 2016, age 41 | Ischemic stroke | Risk factor: Arterial hypertension, grade I; Heart enlargement described on chest X-ray; No specific follow-up recommended. | Clopidogrel 75 mg Piracetam 2400 mg Irbesartan 75 mg |
| July 2020, age 44 | A first episode of persistent atrial fibrillation (AF) registered | Anticoagulant treatment initiated and planned for transesophageal echocardiography (TEE) and electrical cardioversion (ECV). | Dabigatran 2 × 150 mg added |
| August 2020, age 44 | TEE visualized thrombosis of the left atrial appendage (LAA) | Dabigatran switched to Acenocoumarol | |
| September 2020, age 45 | TEE negative for thrombosis of the LAA | Successful ECV | Acenocoumarol continued; Bisoprolol 5 mg as an anti-recurrence treatment |
| March 2021, age 45 | First episode of decompensated heart failure, NYHA III-IV class; A new episode of persistent AF, duration longer than two months | Management of failure according to guidelines; TEE not performed due to INR not being in target; Asymmetric left ventricular hypertrophy and reduced ejection fraction noted; Further diagnostic work-up recommended—cardiac magnetic resonance (CMR) and genetic testing; Test for Anderson–Fabry disease—negative; CMR study positive for cardiomyopathy with diffuse fibrosis, possibly hypertrophic or infiltrative. | At admission: Amiodarone 400 mg; Torasemide 20 mg; Irbesartan 75 mg; Acenocoumarol; At discharge: Torasemide 50 mg; Spironolactone 50 mg; Acenocoumarol; Bisoprolol 5 mg; Sacubitril/valsartan 2 × 24/26 mg; Allopurinol 150 mg; Dapagliflozin 10 mg |
| April 2021 | Compensated state of heart failure Ejection fraction improved to 45% | Selective coronary angiography—negative for epicardial vessel disease, but slow-flow phenomenon present in all coronary arteries TEE revealed LAA thrombosis while in INR therapeutic interval | |
| From 2021 and onwards | Compensated, greatly improved functional capacity Ejection fraction 50% | Refuses pulmonary vein isolation for atrial fibrillation | Torasemide reduced to 10 mg and then to 5 mg |
| March 2023 | Molecular genetic testing revealed three variants, two of which consistent with hypertrophic cardiomyopathy and one possibly related to familial atrial fibrillation. Cascade screening recommended |
| LA A-P Diameter | LA Volume | Ind. LA A-P | LAVI | RA Diameters | RA Area | RA Volume | Ind. RA Volume |
|---|---|---|---|---|---|---|---|
| 56 mm | 130 mL | 22.2 mm/m2 | 52 mL/m2 | 40 × 59 mm | 24 cm2 | 65 mL | 26 mL/m2 |
| IVSd | PWLVd | IVSd/PWLVd | LV EDD | LV ESD | LV EDV | LV ESV | LVEF |
| 16–17 mm; | 8 mm; | 2 | 48 mm | 40 mm | 163 mL | 98 mL | 35–40% |
| LV GLS | PSD | MV E-wave | e’sept. | e’lat. | E/e’ ratio | TAPSE | IVC |
| 12.2% | 57.2 ms | 1.01 m/s | 0.06 m/s | 0.08 m/s | 14.42 | 16 mm | 20 mm, no collapse |
| RV basal diameter | RV EDA | RV ESA | RV FAC | RVFW | PA AT | TR Vmax | Kinetics |
| 41 mm | 21.4 cm2 | 14 cm2 | 35% | 4 mm | 148 ms | 2.2 m/s | Diffuse hypokinesia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gencheva, D.; Angelova, P.; Genova, K.; Atemin, S.; Sleptsova, M.; Todorov, T.; Nikolov, F.; Ruseva, D.; Mitev, V.; Todorova, A. A Cautionary Tale of Hypertrophic Cardiomyopathy—From “Benign” Left Ventricular Hypertrophy to Stroke, Atrial Fibrillation, and Molecular Genetic Diagnostics: A Case Report and Review of Literature. Int. J. Mol. Sci. 2024, 25, 9385. https://doi.org/10.3390/ijms25179385
Gencheva D, Angelova P, Genova K, Atemin S, Sleptsova M, Todorov T, Nikolov F, Ruseva D, Mitev V, Todorova A. A Cautionary Tale of Hypertrophic Cardiomyopathy—From “Benign” Left Ventricular Hypertrophy to Stroke, Atrial Fibrillation, and Molecular Genetic Diagnostics: A Case Report and Review of Literature. International Journal of Molecular Sciences. 2024; 25(17):9385. https://doi.org/10.3390/ijms25179385
Chicago/Turabian StyleGencheva, Dolina, Petya Angelova, Kameliya Genova, Slavena Atemin, Mila Sleptsova, Tihomir Todorov, Fedya Nikolov, Donka Ruseva, Vanyo Mitev, and Albena Todorova. 2024. "A Cautionary Tale of Hypertrophic Cardiomyopathy—From “Benign” Left Ventricular Hypertrophy to Stroke, Atrial Fibrillation, and Molecular Genetic Diagnostics: A Case Report and Review of Literature" International Journal of Molecular Sciences 25, no. 17: 9385. https://doi.org/10.3390/ijms25179385