
Interdependence between Nuclear Pore Gatekeepers and Genome Caretakers: Cues from Genome Instability Syndromes



Abstract
:1. Scope of the Review
2. Architecture of the Nuclear Pore Complex
3. Functions of the Nuclear Pore Complex
4. The NPC and Human Disease
5. Bridging Genome Instability Syndromes to the Nuclear Pore
6. NUP98 Biallelic Variants Underlie a Rothmund–Thomson-like Phenotype
7. NUP98: A Dynamic Multitasking FG-NUP
8. Link of WRN/WRNIP Complex to Nucleoporins
9. Interactions of FAM111B Protease, Mutated in Hereditary Fibrosing Poikiloderma, and Nuclear Pore Components
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Online Mendelian Inheritance in Man (OMIM). Available online: https://www.omim.org/ (accessed on 30 July 2024).
- Colombo, E.A.; Valiante, M.; Uggeri, M.; Orro, A.; Majore, S.; Grammatico, P.; Gentilini, D.; Finelli, P.; Gervasini, C.; D’Ursi, P.; et al. Germline NUP98 Variants in Two Siblings with a Rothmund-Thomson-Like Spectrum: Protein Functional Changes Predicted by Molecular Modeling. Int. J. Mol. Sci. 2023, 24, 4028. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, Y.; Zhai, Y.; Castroagudin, M.R.; Bao, Y.; White, T.E.; Glavy, J.S. Werner complex deficiency in cells disrupts the Nuclear Pore Complex and the distribution of lamin B1. Biochim. Biophys. Acta 2013, 1833, 3338–3345. [Google Scholar] [CrossRef] [PubMed]
- Kliszczak, M.; Moralli, D.; Jankowska, J.D.; Bryjka, P.; Subha Meem, L.; Goncalves, T.; Hester, S.S.; Fischer, R.; Clynes, D.; Green, C.M. Loss of FAM111B protease mutated in hereditary fibrosing poikiloderma negatively regulates telomere length. Front. Cell Dev. Biol. 2023, 11, 1175069. [Google Scholar] [CrossRef]
- Nagai, S.; Dubrana, K.; Tsai-Pflugfelder, M.; Davidson, M.B.; Roberts, T.M.; Brown, G.W.; Varela, E.; Hediger, F.; Gasser, S.M.; Krogan, N.J. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 2008, 322, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.H.; Hoelz, A. The Structure of the Nuclear Pore Complex (an Update). Annu. Rev. Biochem. 2019, 88, 725–783. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sousa, J.; Pereira, C.D.; da Cruz E Silva, O.A.B.; Rebelo, S. Nuclear envelope dysfunction and its contribution to the aging process. Aging Cell 2020, 19, e13143. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, L.; Chen, L.; Gong, B.; Jia, D.; Sun, Q. Nuclear transport proteins: Structure, function, and disease relevance. Signal Transduct. Target Ther. 2023, 8, 425. [Google Scholar] [CrossRef]
- Lima, J.T.; Ferreira, J.G. Mechanobiology of the nucleus during the G2-M transition. Nucleus 2024, 15, 2330947. [Google Scholar] [CrossRef]
- Sun, J.; Shi, Y.; Yildirim, E. The Nuclear Pore Complex in Cell Type-Specific Chromatin Structure and Gene Regulation. Trends Genet. 2019, 35, 579–588. [Google Scholar] [CrossRef]
- Brickner, J.H. The nuclear pore complex as a platform for epigenetic regulation. J. Cell Biol. 2023, 222, e202307078. [Google Scholar] [CrossRef]
- Gaillard, H.; Santos-Pereira, J.M.; Aguilera, A. The Nup84 complex coordinates the DNA damage response to warrant genome integrity. Nucleic Acids Res. 2019, 47, 4054–4067. [Google Scholar] [CrossRef]
- Nobari, P.; Doye, V.; Boumendil, C. Metazoan nuclear pore complexes in gene regulation and genome stability. DNA Repair 2023, 130, 103565. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.N.; Dubrana, K.; Palancade, B. On the edge: How nuclear pore complexes rule genome stability. Curr. Opin. Genet. Dev. 2024, 84, 102150. [Google Scholar] [CrossRef]
- Buxboim, A.; Kronenberg-Tenga, R.; Salajkova, S.; Avidan, N.; Shahak, H.; Thurston, A.; Medalia, O. Scaffold, mechanics and functions of nuclear lamins. FEBS Lett. 2023, 597, 2791–2805. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 67, 2215–2230. [Google Scholar] [CrossRef]
- Hampoelz, B.; Andres-Pons, A.; Kastritis, P.; Beck, M. Structure and Assembly of the Nuclear Pore Complex. Annu. Rev. Biophys. 2019, 48, 515–536. [Google Scholar] [CrossRef]
- Bley, C.J.; Nie, S.; Mobbs, G.W.; Petrovic, S.; Gres, A.T.; Liu, X.; Mukherjee, S.; Harvey, S.; Huber, F.M.; Lin, D.H.; et al. Architecture of the cytoplasmic face of the nuclear pore. Science 2022, 376, eabm9129. [Google Scholar] [CrossRef]
- Mosalaganti, S.; Obarska-Kosinska, A.; Siggel, M.; Taniguchi, R.; Turoňová, B.; Zimmerli, C.E.; Buczak, K.; Schmidt, F.H.; Margiotta, E.; Mackmull, M.T.; et al. AI-based structure prediction empowers integrative structural analysis of human nuclear pores. Science 2022, 376, eabm9506. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, S.; Samanta, D.; Perriches, T.; Bley, C.J.; Thierbach, K.; Brown, B.; Nie, S.; Mobbs, G.W.; Stevens, T.A.; Liu, X.; et al. Architecture of the linker-scaffold in the nuclear pore. Science 2022, 376, eabm9798. [Google Scholar] [CrossRef]
- Biorender. Available online: https://www.biorender.com/ (accessed on 30 July 2024).
- Lyngdoh, D.L.; Nag, N.; Uversky, V.N.; Tripathi, T. Prevalence and functionality of intrinsic disorder in human FG-nucleoporins. Int. J. Biol. Macromol. 2021, 175, 156–170. [Google Scholar] [CrossRef]
- Huang, G.; Zeng, C.; Shi, Y. Structure of the nuclear pore complex goes atomic. Curr. Opin. Struct. Biol. 2023, 78, 102523. [Google Scholar] [CrossRef]
- Fontana, P.; Dong, Y.; Pi, X.; Tong, A.B.; Hecksel, C.W.; Wang, L.; Fu, T.M.; Bustamante, C.; Wu, H. Structure of cytoplasmic ring of nuclear pore complex by integrative cryo-EM and AlphaFold. Science 2022, 376, eabm9326. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.; Richter, R.P.; Gorlich, D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science 2006, 314, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.; Görlich, D. A saturated FG-repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell 2007, 130, 512–523. [Google Scholar] [CrossRef]
- Milles, S.; Mercadante, D.; Aramburu, I.V.; Jensen, M.R.; Banterle, N.; Koehler, C.; Tyagi, S.; Clarke, J.; Shammas, S.L.; Blackledge, M.; et al. Plasticity of an ultrafast interaction between nucleoporins and nuclear transport receptors. Cell 2015, 163, 734–745. [Google Scholar] [CrossRef]
- Wing, C.E.; Fung, H.Y.J.; Chook, Y.M. Karyopherin-mediated nucleocytoplasmic transport. Nat. Rev. Mol. Cell Biol. 2022, 23, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Kehlenbach, R.H.; Neumann, P.; Ficner, R.; Dickmanns, A. Interaction of nucleoporins with nuclear transport receptors: A structural perspective. Biol. Chem. 2023, 404, 791–805. [Google Scholar] [CrossRef]
- Ren, Y.; Seo, H.S.; Blobel, G.; Hoelz, A. Structural and functional analysis of the interaction between the nucleoporin Nup98 and the mRNA export factor Rae1. Proc. Natl. Acad. Sci. USA 2010, 107, 10406–10411. [Google Scholar] [CrossRef]
- Fischer, J.; Teimer, R.; Amlacher, S.; Kunze, R.; Hurt, E. Linker Nups connect the nuclear pore complex inner ring with the outer ring and transport channel. Nat. Struct. Mol. Biol. 2015, 22, 774–781. [Google Scholar] [CrossRef]
- Zimmerli, C.E.; Allegretti, M.; Rantos, V.; Goetz, S.K.; Obarska-Kosinska, A.; Zagoriy, I.; Halavatyi, A.; Hummer, G.; Mahamid, J.; Kosinski, J.; et al. Nuclear pores dilate and constrict in cellulo. Science 2021, 374, eabd9776. [Google Scholar] [CrossRef]
- Matsuda, A.; Mofrad, M.R.K. On the nuclear pore complex and its emerging role in cellular mechanotransduction. APL Bioeng. 2022, 6, 011504. [Google Scholar] [CrossRef] [PubMed]
- Izaurralde, E.; Kutay, U.; von Kobbe, C.; Mattaj, I.W.; Görlich, D. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J. 1997, 16, 6535–6547. [Google Scholar] [CrossRef]
- Yu, M.; Heidari, M.; Mikhaleva, S.; Tan, P.S.; Mingu, S.; Ruan, H.; Reinkemeier, C.D.; Obarska-Kosinska, A.; Siggel, M.; Beck, M.; et al. Visualizing the disordered nuclear transport machinery in situ. Nature 2023, 617, 162–169. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.A. Nuclear pore complexes as hubs for gene regulation. Nucleus 2017, 9, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Garcia, P.; Capelson, M. The nuclear pore complex and the genome: Organizing and regulatory principles. Curr. Opin. Genet. Dev. 2021, 67, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Lamm, N.; Rogers, S.; Cesare, A.J. Chromatin mobility and relocation in DNA repair. Trends Cell Biol. 2021, 31, 843–855. [Google Scholar] [CrossRef]
- Mojumdar, A.; Mair, N.; Adam, N.; Cobb, J.A. Changes in DNA double-strand break repair during aging correlate with an increase in genomic mutations. J. Mol. Biol. 2022, 434, 167798. [Google Scholar] [CrossRef]
- Sakuma, S.; D’Angelo, M.A. The roles of the nuclear pore complex in cellular dysfunction, aging and disease. Semin. Cell Dev. Biol. 2017, 68, 72–84. [Google Scholar] [CrossRef]
- Shamanna, R.A.; Croteau, D.L.; Lee, J.H.; Bohr, V.A. Recent Advances in Understanding Werner Syndrome. F1000Res 2017, 6, 1779. [Google Scholar] [CrossRef]
- Lu, L.; Jin, W.; Wang, L.L. Aging in Rothmund-Thomson syndrome and related RECQL4 genetic disorders. Ageing Res. Rev. 2017, 33, 30–35. [Google Scholar] [CrossRef]
- Milosic, F.; Hengstschläger, M.; Osmanagic-Myers, S. Premature aging in genetic diseases: What conclusions can be drawn for physiological aging. Front. Aging 2024, 4, 1327833. [Google Scholar] [CrossRef]
- Jühlen, R.; Fahrenkrog, B. From the sideline: Tissue-specific nucleoporin function in health and disease, an update. FEBS Lett. 2023, 597, 2750–2768. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef] [PubMed]
- Nofrini, V.; Di Giacomo, D.; Mecucci, C. Nucleoporin genes in human diseases. Eur. J. Hum. Genet. 2016, 24, 1388–1395. [Google Scholar] [CrossRef]
- Michmerhuizen, N.L.; Klco, J.M.; Mullighan, C.G. Mechanistic insights and potential therapeutic approaches for NUP98-rearranged hematologic malignancies. Blood 2020, 136, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Davis, E.S.; Daugird, T.A.; Zhao, S.; Quiroga, I.Y.; Uryu, H.; Li, J.; Storey, A.J.; Tsai, Y.H.; Keeley, D.P.; et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 2021, 595, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Brindle, P.K.; Schnabel, C.A.; Pritchard, C.E.; Cleary, M.L.; van Deursen, J.M. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol. Cell. Biol. 1999, 19, 764–776. [Google Scholar] [CrossRef]
- Oka, M.; Otani, M.; Miyamoto, Y.; Oshima, R.; Adachi, J.; Tomonaga, T.; Asally, M.; Nagaoka, Y.; Tanaka, K.; Toyoda, A.; et al. Phase-separated nuclear bodies of nucleoporin fusions promote condensation of MLL1/CRM1 and rearrangement of 3D genome structure. Cell Rep. 2023, 42, 112884. [Google Scholar] [CrossRef]
- Coyne, A.N.; Rothstein, J.D. Nuclear pore complexes—A doorway to neural injury in neurodegeneration. Nat. Rev. Neurol. 2022, 18, 348–362. [Google Scholar] [CrossRef]
- Dickson, J.R.; Frosch, M.P.; Hyman, B.T. Altered localization of nucleoporin 98 in primary tauopathies. Brain Commun. 2022, 5, fcac334. [Google Scholar] [CrossRef]
- Nag, N.; Tripathi, T. Tau-FG nucleoporin98 interaction and impaired nucleocytoplasmic transport in Alzheimer’s disease. Brief. Funct. Genom. 2022, 22, 161–167. [Google Scholar] [CrossRef]
- Kumar, M.S.; Stallworth, K.M.; Murthy, A.C.; Lim, S.M.; Li, N.; Jain, A.; Munro, J.B.; Fawzi, N.L.; Lagier-Tourenne, C.; Bosco, D.A. Interactions between FUS and the C-terminal Domain of NUP62 are Sufficient for their Co-phase Separation into Amorphous Assemblies. J. Mol. Biol. 2023, 435, 167972. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Baskerville, V.; Zaepfel, B.L.; Dickson, D.W.; Rigo, F.; Bennett, F.; Lusk, C.P.; Rothstein, J.D. Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci. Transl. Med. 2021, 13, eabe1923. [Google Scholar] [CrossRef]
- Wang, L.L.; Plon, S.E. Rothmund-Thomson Syndrome. In GeneReviews; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2022; pp. 1993–2022. [Google Scholar]
- Ajeawung, N.F.; Nguyen, T.T.M.; Lu, L.; Kucharski, T.J.; Rousseau, J.; Molidperee, S.; Atienza, J.; Gamache, I.; Jin, W.; Plon, S.E.; et al. Mutations in ANAPC1, Encoding a Scaffold Subunit of the Anaphase-Promoting Complex, Cause Rothmund-Thomson Syndrome Type 1. Am. J. Hum. Genet. 2019, 105, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, K.B.; Malureanu, L.; van Deursen, J.M. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature 2005, 438, 1036–1039. [Google Scholar] [CrossRef]
- Luong, T.T.; Bernstein, K.A. Role and Regulation of the RECQL4 Family during Genomic Integrity Maintenance. Genes 2021, 12, 1919. [Google Scholar] [CrossRef] [PubMed]
- Oshima, J.; Martin, G.M.; Hisama, F.M. Werner Syndrome. In GeneReviews; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2021; pp. 1993–2022. [Google Scholar]
- Lu, H.; Shamanna, R.A.; de Freitas, J.K.; Okur, M.; Khadka, P.; Kulikowicz, T.; Holland, P.P.; Tian, J.; Croteau, D.L.; Davis, A.J.; et al. Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat. Commun. 2017, 8, 2039. [Google Scholar] [CrossRef]
- Lee, J.H.; Shamanna, R.A.; Kulikowicz, T.; Borhan Fakouri, N.; Kim, E.W.; Christiansen, L.S.; Croteau, D.L.; Bohr, V.A. CDK2 phosphorylation of Werner protein (WRN) contributes to WRN’s DNA double-strand break repair pathway choice. Aging Cell 2021, 20, e13484. [Google Scholar] [CrossRef]
- Palancade, B.; Liu, X.; Garcia-Rubio, M.; Aguilera, A.; Zhao, X.; Doye, V. Nucleoporins prevent DNA damage accumulation by modulating Ulp1-dependent sumoylation processes. Mol. Biol. Cell 2007, 18, 2912–2923. [Google Scholar] [CrossRef]
- Kaur, S.; White, T.E.; Di Guilio, A.L.; Glavy, J.S. The discovery of a Werner Helicase Interacting Protein (WHIP) association with the nuclear pore complex. Cell Cycle 2010, 9, 3106–3111. [Google Scholar] [CrossRef]
- Mercier, S.; Küry, S.; Shaboodien, G.; Houniet, D.T.; Khumalo, N.P.; Bou-Hanna, C.; Bodak, N.; Cormier-Daire, V.; David, A.; Faivre, L.; et al. Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am. J. Hum. Genet. 2013, 93, 1100–1107. [Google Scholar] [CrossRef]
- Mercier, S.; Küry, S.; Barbarot, S. Hereditary Fibrosing Poikiloderma with Tendon Contractures, Myopathy, and Pulmonary Fibrosis. In GeneReviews; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2016; pp. 1993–2024. [Google Scholar]
- ENSEMBL. Available online: https://www.ensembl.org/index.html (accessed on 10 July 2024).
- GNOMAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 10 July 2024).
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Jühlen, R.; Fahrenkrog, B. Moonlighting nuclear pore proteins: Tissue-specific nucleoporin function in health and disease. Histochem. Cell Biol. 2018, 150, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Garcia, P.; Debo, B.; Aleman, J.R.; Talamas, J.A.; Lan, Y.; Nguyen, N.H.; Won, K.J.; Capelson, M. Metazoan Nuclear Pores Provide a Scaffold for Poised Genes and Mediate Induced Enhancer-Promoter Contacts. Mol. Cell 2017, 66, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Garcia, P.; Capelson, M. Nuclear pores in genome architecture and enhancer function. Curr. Opin. Cell Biol. 2019, 58, 126–133. [Google Scholar] [CrossRef]
- Wu, G.; Glickstein, S.; Liu, W.; Fujita, T.; Li, W.; Yang, Q.; Duvoisin, R.; Wan, Y. The anaphase-promoting complex coordinates initiation of lens differentiation. Mol. Biol. Cell 2007, 18, 1018–1029. [Google Scholar] [CrossRef]
- Marabitti, V.; Lillo, G.; Malacaria, E.; Palermo, V.; Sanchez, M.; Pichierri, P.; Franchitto, A. ATM pathway activation limits R-loop-associated genomic instability in Werner syndrome cells. Nucleic Acids Res. 2019, 47, 3485–3502. [Google Scholar] [CrossRef]
- Larizza, L.; Roversi, G.; Volpi, L. Rothmund-Thomson syndrome. Orphanet. J. Rare Dis. 2010, 5, 2. [Google Scholar] [CrossRef]
- Bilir, Ş.; Kojidani, T.; Mori, C.; Osakada, H.; Kobayashi, S.; Koujin, T.; Hiraoka, Y.; Haraguchi, T. Roles of NUP133, NUP153 and membrane fenestrations in assembly of the nuclear pore complex at the end of mitosis. Genes Cells 2019, 24, 338–353. [Google Scholar] [CrossRef]
- Ibáñez de Opakua, A.; Geraets, J.A.; Frieg, B.; Dienemann, C.; Savastano, A.; Rankovic, M.; Cima-Omori, M.S.; Schröder, G.F.; Zweckstetter, M. Molecular interactions of FG nucleoporin repeats at high resolution. Nat. Chem. 2022, 14, 1278–1285. [Google Scholar] [CrossRef]
- Ori, A.; Banterle, N.; Iskar, M.; Andrés-Pons, A.; Escher, C.; Khanh Bui, H.; Sparks, L.; Solis-Mezarino, V.; Rinner, O.; Bork, P.; et al. Cell type-specific nuclear pores: A case in point for context-dependent stoichiometry of molecular machines. Mol. Syst. Biol. 2013, 9, 648. [Google Scholar] [CrossRef]
- De Boer, H.R.; Guerrero Llobet, S.; van Vugt, M.A. Controlling the response to DNA damage by the APC/C-Cdh1. Cell. Mol. Life Sci. 2016, 73, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Fontoura, B.M.A.; Blobel, G.; Matunis, M.J. A conserved biogenesis pathway for nucleoporins: Proteolytic processing of a 186-kilodalton precursor generates Nup98 and the novel nucleoporin, Nup96. J. Cell Biol. 1999, 144, 1097–1112. [Google Scholar] [CrossRef]
- Naylor, R.M.; Jeganathan, K.B.; Cao, X.; van Deursen, J.M. Nuclear pore protein NUP88 activates anaphase-promoting complex to promote aneuploidy. J. Clin. Investig. 2016, 126, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Laurell, E.; Beck, K.; Krupina, K.; Theerthagiri, G.; Bodenmiller, B.; Horvath, P.; Aebersold, R.; Antonin, W.; Kutay, U. Phosphorylation of Nup98 by multiple kinases is crucial for NPC disassembly during mitotic entry. Cell 2011, 144, 539–550. [Google Scholar] [CrossRef]
- Schmidt, H.B.; Görlich, D. NUP98 FG domains from diverse species spontaneously phase-separate into particles with nuclear pore-like permselectivity. Elife 2015, 4, e04251. [Google Scholar] [CrossRef]
- Celetti, G.; Paci, G.; Caria, J.; Van Delinder, V.; Bachand, G.; Lemke, E.A. The liquid state of FG-nucleoporins mimics permeability barrier properties of nuclear pore complexes. J. Cell Biol. 2020, 219, e201907157. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.; Van der Giessen, E.; Onck, P.R. Phase separation of intrinsically disordered FG-Nups is driven by highly dynamic FG motifs. Proc. Natl. Acad. Sci. USA 2023, 120, e2221804120. [Google Scholar] [CrossRef]
- Oka, M.; Mura, S.; Yamada, K.; Sangel, P.; Hirata, S.; Maehara, K.; Kawakami, K.; Tachibana, T.; Ohkawa, Y.; Kimura, H.; et al. Chromatin-prebound Crm1 recruits Nup98-HoxA9 fusion to induce aberrant expression of Hox cluster genes. Elife 2016, 5, e09540. [Google Scholar] [CrossRef]
- Bertrums, E.J.M.; Smith, J.L.; Harmon, L.; Ries, R.E.; Wang, Y.J.; Alonzo, T.A.; Menssen, A.J.; Chisholm, K.M.; Leonti, A.R.; Tarlock, K.; et al. Comprehensive molecular and clinical characterization of NUP98 fusions in pediatric acute myeloid leukemia. Haematologica 2023, 108, 2044–2058. [Google Scholar] [CrossRef]
- Ropert, B.; Gallrein, C.; Schumacher, B. DNA repair deficiencies and neurodegeneration. DNA Repair 2024, 138, 103679. [Google Scholar] [CrossRef]
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA.; Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Turaga, R.V.; Paquet, E.R.; Sild, M.; Vignard, J.; Garand, C.; Johnson, F.B.; Masson, J.Y.; Lebel, M. The Werner syndrome protein affects the expression of genes involved in adipogenesis and inflammation in addition to cell cycle and DNA damage responses. Cell Cycle 2009, 8, 2080–2092. [Google Scholar] [CrossRef]
- Crabbe, L.; Jauch, A.; Naeger, C.M.; Holtgreve-Grez, H.; Karlseder, J. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 2205–2210. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, Y.; Seki, M.; Yoshimura, A.; Nishino, K.; Hayashi, T.; Takeuchi, T.; Iguchi, S.; Kusa, Y.; Ohtsuki, M.; Tsuyama, T.; et al. Analyses of the interaction of WRNIP1 with Werner syndrome protein (WRN) in vitro and in the cell. DNA Repair 2006, 5, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Seki, M.; Enomoto, T. The role of WRNIP1 in genome maintenance. Cell Cycle 2017, 16, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.R.; Zhai, Y.; Li, Z.; Zhao, H.; Mackmull, M.T.; Glavy, J.S. Discovering the nuclear localization signal of Werner Helicase Interacting Protein 1. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119502. [Google Scholar] [CrossRef]
- Zuccolo, M.; Alves, A.; Galy, V.; Bolhy, S.; Formstecher, E.; Racine, V.; Sibarita, J.B.; Fukagawa, T.; Shiekhattar, R.; Yen, T.; et al. The human NUP107-160 nuclear pore subcomplex contributes to proper kinetochore functions. EMBO J. 2007, 26, 1853–1864. [Google Scholar] [CrossRef]
- Mansfeld, J.; Güttinger, S.; Hawryluk-Gara, L.A.; Panté, N.; Mall, M.; Galy, V.; Haselmann, U.; Mühlhäusser, P.; Wozniak, R.W.; Mattaj, I.W.; et al. The conserved transmembrane nucleoporin NDC1 is required for nuclear pore complex assembly in vertebrate cells. Mol. Cell 2006, 22, 93–103. [Google Scholar] [CrossRef]
- Stavru, F.; Hülsmann, B.B.; Spang, A.; Hartmann, E.; Cordes, V.C.; Görlich, D. NDC1: A crucial membrane-integral nucleoporin of metazoan nuclear pore complexes. J. Cell Biol. 2006, 173, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.L.; Roux, K.J.; Burke, B. Blurring the boundary: The nuclear envelope extends its reach. Science 2007, 318, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Lachapelle, S.; Gagné, J.P.; Garand, C.; Desbiens, M.; Coulombe, Y.; Bohr, V.A.; Hendzel, M.J.; Masson, J.Y.; Poirier, G.G.; Lebel, M. Proteome-wide identification of WRN-interacting proteins in untreated and nuclease-treated samples. J. Proteome Res. 2011, 10, 1216–1227. [Google Scholar] [CrossRef]
- Marciniak, R.A.; Lombard, D.B.; Johnson, F.B.; Guarente, L. Nucleolar localization of the Werner syndrome protein in human cells. Proc. Natl. Acad. Sci. USA 1998, 95, 6887–6892. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.B.; Datta, S.; Snow, C.J.; Chatterjee, M.; Ni, L.; Spencer, A.; Yang, C.S.; Cubeñas-Potts, C.; Matunis, M.J.; Paschal, B.M. The defective nuclear lamina in Hutchinson-Gilford progeria syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Mol. Cell Biol. 2011, 31, 3378–3395. [Google Scholar] [CrossRef] [PubMed]
- Khumalo, N.P.; Pillay, K.; Beighton, P.; Wainwright, H.; Walker, B.; Saxe, N.; Mayosi, B.M.; Bateman, E.D. Poikiloderma.; tendon contracture and pulmonary fibrosis: A new autosomal dominant syndrome? Br. J. Dermatol. 2006, 155, 1057–1061. [Google Scholar] [CrossRef]
- Hoffmann, S.; Pentakota, S.; Mund, A.; Haahr, P.; Coscia, F.; Gallo, M.; Mann, M.; Taylor, N.M.; Mailand, N. FAM111 protease activity undermines cellular fitness and is amplified by gain-of-function mutations in human disease. EMBO Rep. 2020, 21, e50662. [Google Scholar] [CrossRef]
- Nie, M.; Oravcová, M.; Jami-Alahmadi, Y.; Wohlschlegel, J.A.; Lazzerini-Denchi, E.; Boddy, M.N. FAM111A induces nuclear dysfunction in disease and viral restriction. EMBO Rep. 2021, 22, e50803. [Google Scholar] [CrossRef]
- Naicker, D.; Rhoda, C.; Sunda, F.; Arowolo, A. Unravelling the Intricate Roles of FAM111A and FAM111B: From Protease-Mediated Cellular Processes to Disease Implications. Int. J. Mol. Sci. 2024, 25, 2845. [Google Scholar] [CrossRef]
- Cheng, S.; Lo, I.F.M.; Luk, H.M. FAM111A-Related Skeletal Dysplasias. In GeneReviews; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2023; pp. 1993–2024. [Google Scholar]
- Mercier, S.; Küry, S.; Nahon, S.; Salort-Campana, E.; Barbarot, S.; Bézieau, S. FAM111B mutation is associated with pancreatic cancer predisposition. Pancreas 2019, 48, e41–e42. [Google Scholar] [CrossRef]
- Arowolo, A.; Rhoda, C.; Khumalo, N. Mutations within the putative protease domain of the human FAM111B gene may predict disease severity and poor prognosis: A review of POIKTMP cases. Exp. Dermatol. 2022, 31, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Machida, Y.; Palani, S.; Caulfield, T.R.; Radisky, E.S.; Kaufmann, S.H.; Machida, Y.J. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat. Commun. 2020, 11, 1318. [Google Scholar] [CrossRef] [PubMed]
- Roversi, G.; Colombo, E.A.; Magnani, I.; Gervasini, C.; Maggiore, G.; Paradisi, M.; Larizza, L. Spontaneous chromosomal instability in peripheral blood lymphocytes from two molecularly confirmed Italian patients with Hereditary Fibrosis Poikiloderma: Insights into cancer predisposition. Genet. Mol. Biol. 2021, 44, e20200332. [Google Scholar] [CrossRef]
- Davey, N.E.; Morgan, D.O. Building a Regulatory Network with Short Linear Sequence Motifs: Lessons from the Degrons of the Anaphase-Promoting Complex. Mol. Cell 2016, 64, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynska, A.; Gonzalo, S.; Shanahan, C.; Askjaer, P. The nuclear lamina in health and disease. Nucleus 2016, 7, 233–248. [Google Scholar] [CrossRef]
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 2020, 19, 464–473. [Google Scholar] [CrossRef]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Rossi, M.L.; Singh, D.K.; Dunn, C.; Ramamoorthy, M.; Croteau, D.L.; Liu, Y.; Bohr, V.A. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J. Biol. Chem. 2012, 287, 196–209. [Google Scholar] [CrossRef]
- Sezer, A.; Kayhan, G.; Gursoy, T.R.; Eyuboglu, T.S.; Percin, F.E. A homozygous missense variant in the WRN gene segregating in a family with progressive pulmonary failure with recurrent spontaneous pneumothorax and interstitial lung disease. Am. J. Med. Genet. A 2023, 191, 220–227. [Google Scholar] [CrossRef]
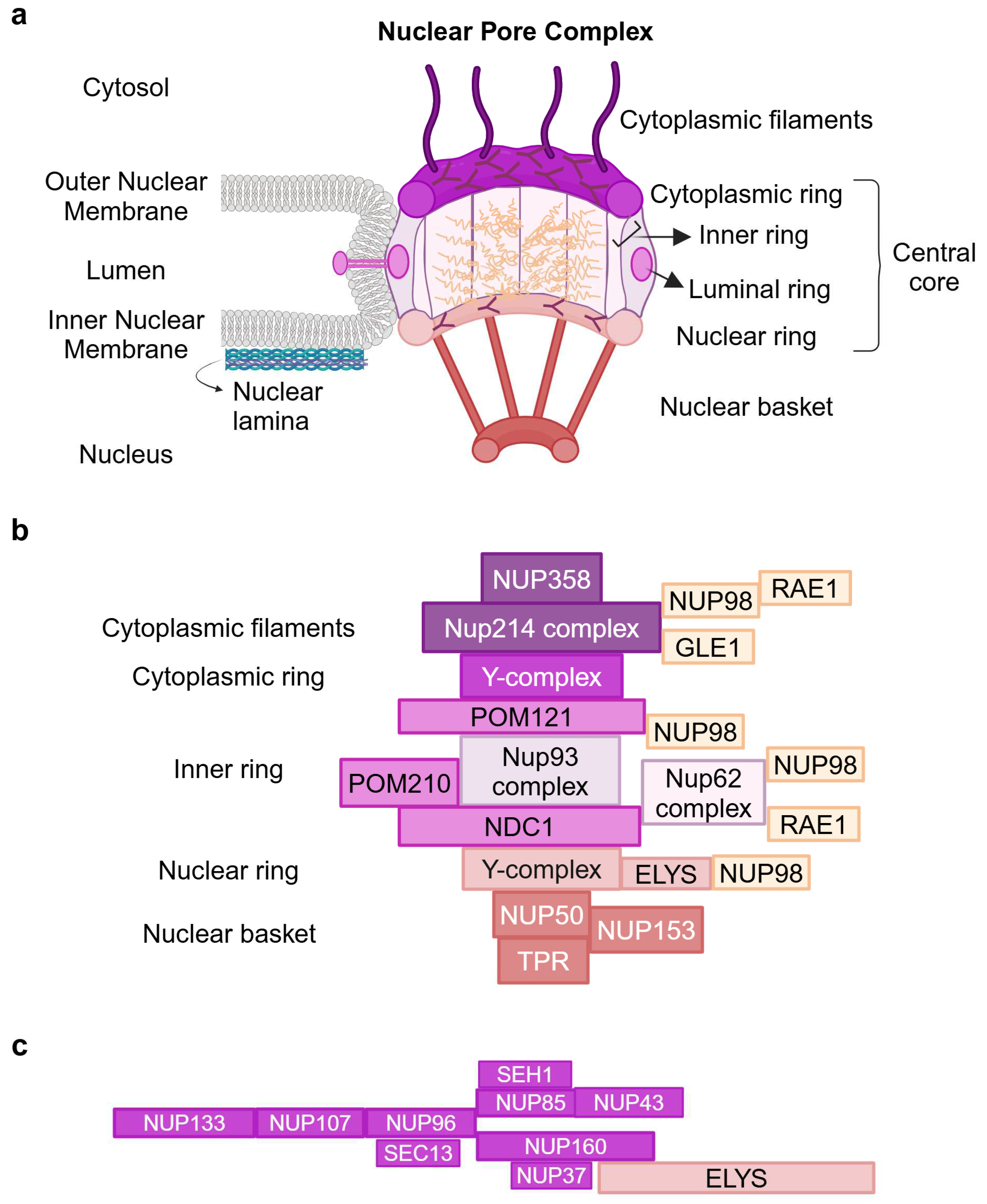
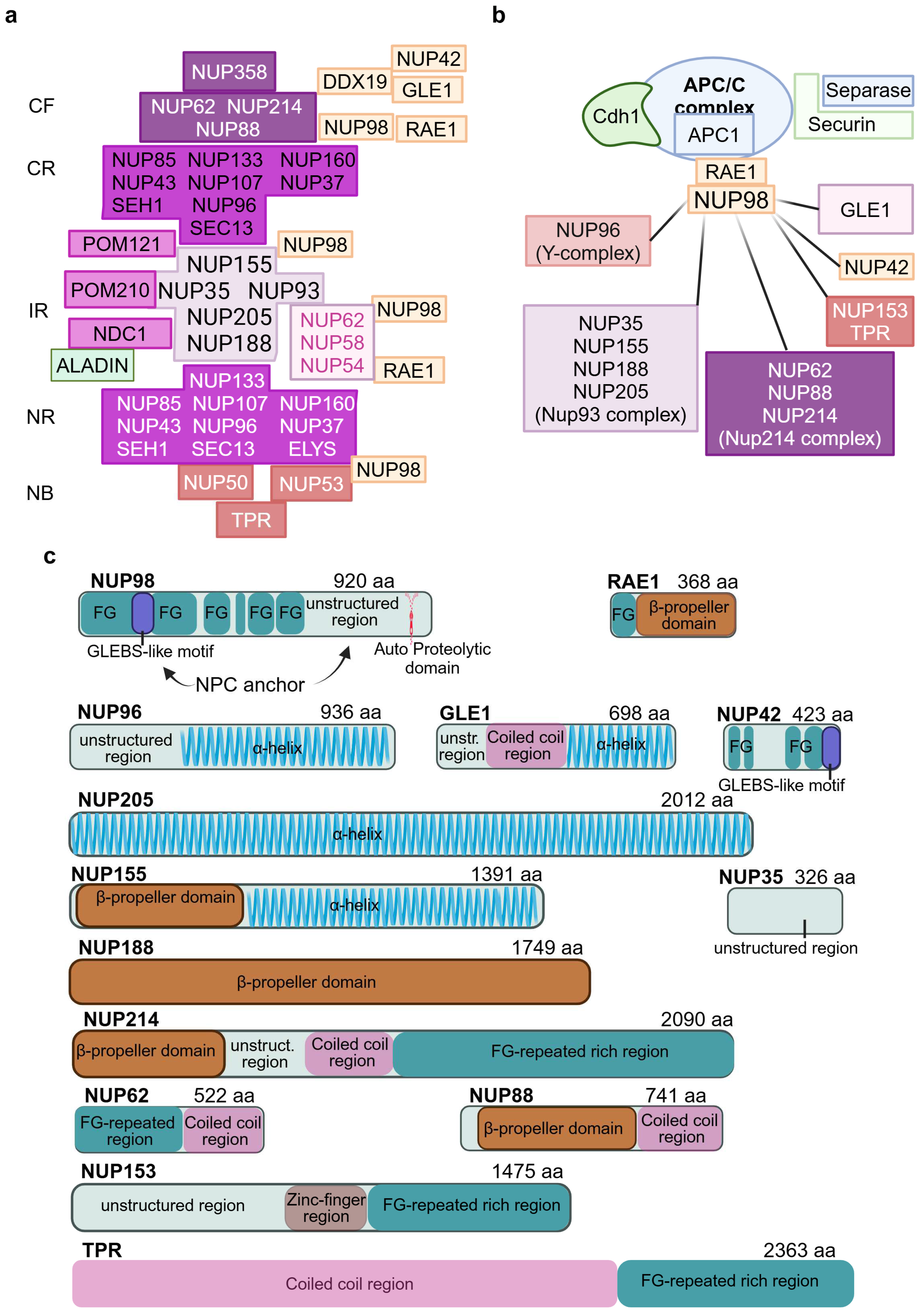



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | NUP98 Siblings | RTS2 | RTS1 |
|---|---|---|---|
| Causative gene | NUP98 | RECQL4 | ANAPC1 |
| Skin pigmentation | Mottled pigmentation since adolescence | Early poikiloderma | Early poikiloderma |
| Dry and fragile skin | + | + | + |
| Thin and fragile hair | + | + | + |
| Eyelashes/eyebrows | Absent | Absent | Absent |
| Eye abnormalities | Bilateral cataracts since infancy | − | Juvenile cataracts |
| Ungual abnormalities | − | + | ± |
| Dental decay | + since adolescence | + | ± |
| Short stature | + * | + | + |
| Skeletal abnormalities | Osteoporosis in third decade | + | + |
| Hypogonadism/oligomenorrhea | + | +/− | + |
| Malignancies | − (untill sixth decade) | Cancer predisposition Osteosarcoma/skin cancer | ? |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larizza, L.; Colombo, E.A. Interdependence between Nuclear Pore Gatekeepers and Genome Caretakers: Cues from Genome Instability Syndromes. Int. J. Mol. Sci. 2024, 25, 9387. https://doi.org/10.3390/ijms25179387
Larizza L, Colombo EA. Interdependence between Nuclear Pore Gatekeepers and Genome Caretakers: Cues from Genome Instability Syndromes. International Journal of Molecular Sciences. 2024; 25(17):9387. https://doi.org/10.3390/ijms25179387
Chicago/Turabian StyleLarizza, Lidia, and Elisa Adele Colombo. 2024. "Interdependence between Nuclear Pore Gatekeepers and Genome Caretakers: Cues from Genome Instability Syndromes" International Journal of Molecular Sciences 25, no. 17: 9387. https://doi.org/10.3390/ijms25179387