
An Interplay between Transcription Factors and Recombinant Protein Synthesis in Yarrowia lipolytica at Transcriptional and Functional Levels—The Global View


Abstract
:1. Introduction
2. Results and Discussion
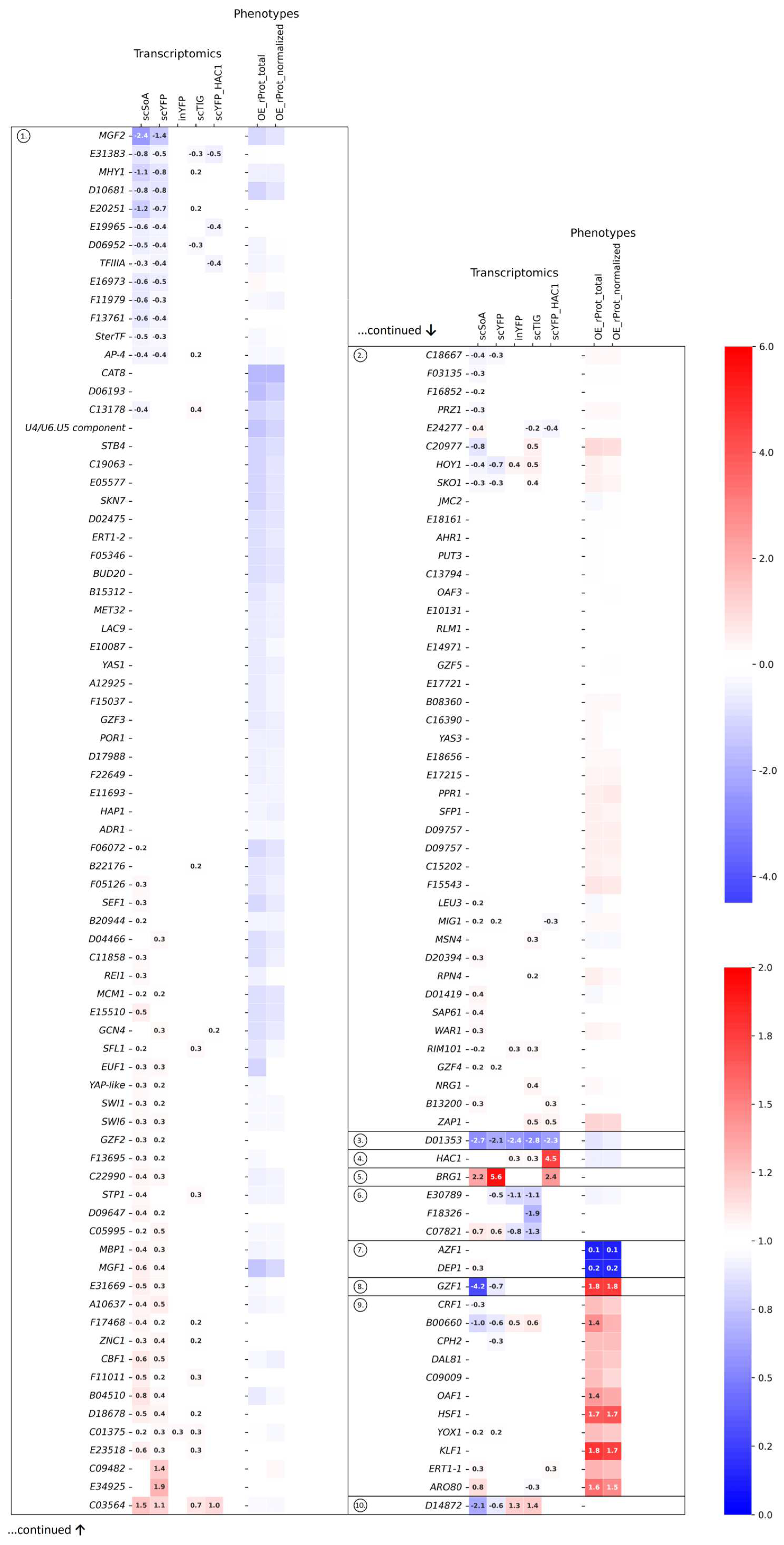
2.1. Data Extraction, Juxtaposition, and Global Clustering—Clusters Overview
2.2. Statistical Overrepresentation Test Delimited Two Major Categories amongst TFs
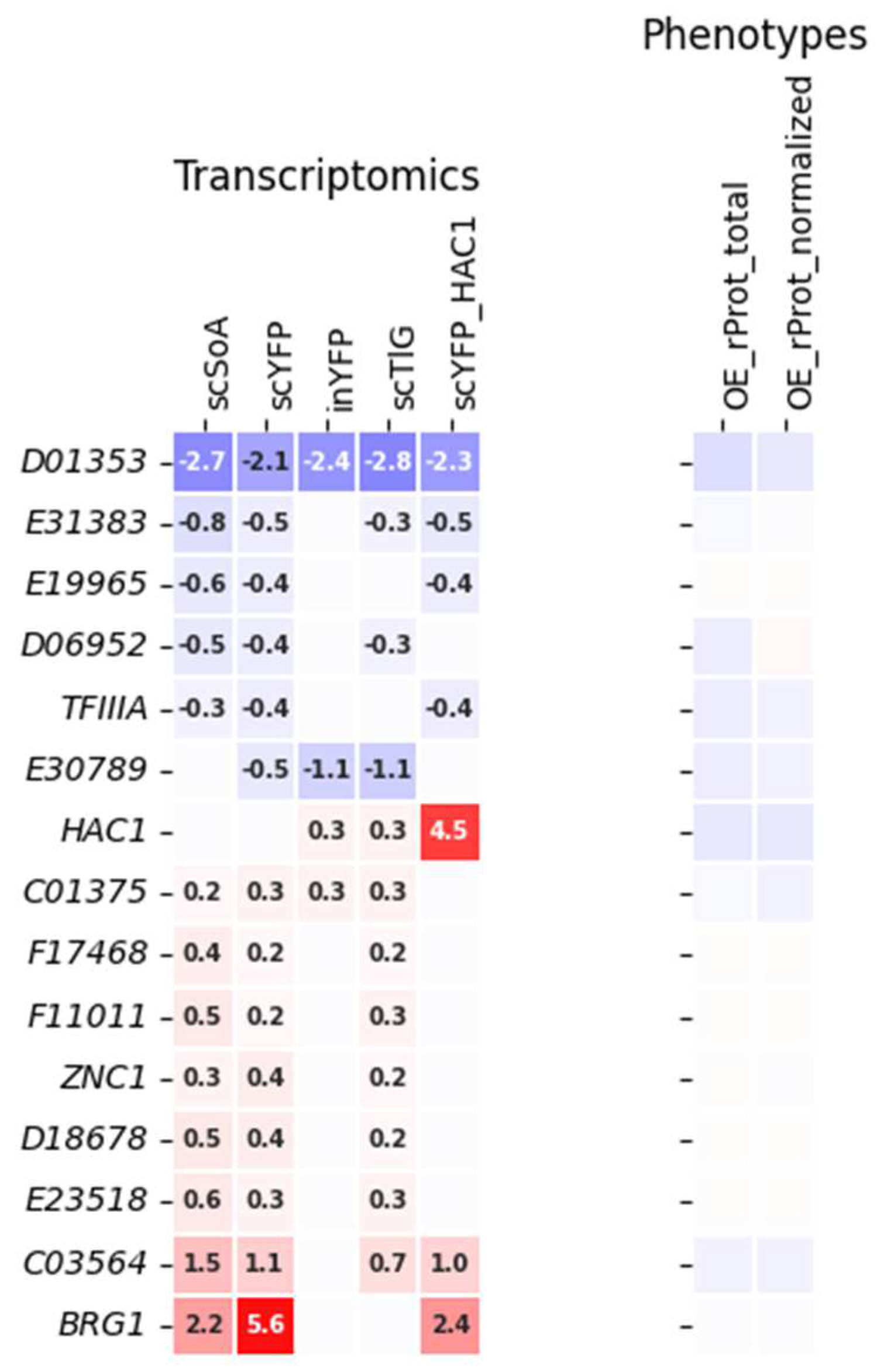
2.3. Usefulness of Specific Deregulation Patterns as Selectors of TFs Involved in Specific Biological Processes
2.4. Direct Comparison of Phenotypes Elicited by Y. lipolytica Strains Bearing OE or KO of Selected TFs
3. Summary and Conclusions
4. Materials and Methods
4.1. Microbial Strains Used for Transcriptomics and Functional OE Screens Data Acquisition
4.2. Microbial Strains Used for Functional Studies—KO in a Specific TF loci
Construction of the KO Strains—Deletion Cassette and CRISPR-Cas9 Targeting
4.3. Functional Studies—Direct Comparative Study of OE and KO Strains
4.3.1. Cultivation Conditions
4.3.2. Samples Analysis
4.4. Omics and Functional Data Acquisition
4.5. Data Processing and Mathematical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, B.; Tan, K. Understanding Transcriptional Regulatory Networks using Computational Models. Curr. Opin. Genet. Dev. 2016, 37, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Su, K.; Katebi, A.; Kohar, V.; Clauss, B.; Gordin, D.; Qin, Z.S.; Karuturi, R.K.M.; Li, S.; Lu, M. NetAct: A Computational Platform to Construct Core Transcription Factor Regulatory Networks using Gene Activity. Genome Biol. 2022, 23, 270. [Google Scholar] [CrossRef]
- Czajka, J.J.; Oyetunde, T.; Tang, Y.J. Integrated Knowledge Mining, Genome-Scale Modeling, and Machine Learning for Predicting Yarrowia lipolytica Bioproduction. Metab. Eng. 2021, 67, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Madzak, C. Engineering Yarrowia lipolytica for Use in Biotechnological Applications: A Review of Major Achievements and Recent Innovations. Mol. Biotechnol. 2018, 60, 621–635. [Google Scholar] [CrossRef]
- Madzak, C. Yarrowia lipolytica Strains and Their Biotechnological Applications: How Natural Biodiversity and Metabolic Engineering Could Contribute to Cell Factories Improvement. J. Fungi 2021, 7, 548. [Google Scholar] [CrossRef] [PubMed]
- Celińska, E.; Nicaud, J.-M. Filamentous Fungi-like Secretory Pathway Strayed in a Yeast System: Peculiarities of Yarrowia lipolytica Secretory Pathway Underlying Its Extraordinary Performance. Appl. Microbiol. Biotechnol. 2019, 103, 39–52. [Google Scholar] [CrossRef]
- Theron, C.W.; Vandermies, M.; Telek, S.; Steels, S.; Fickers, P. Comprehensive Comparison of Yarrowia lipolytica and Pichia Pastoris for Production of Candida Antarctica Lipase B. Sci. Rep. 2020, 10, 1741. [Google Scholar] [CrossRef]
- Vandermies, M.; Denies, O.; Nicaud, J.-M.; Fickers, P. EYK1 Encoding Erythrulose Kinase as a Catabolic Selectable Marker for Genome Editing in the Non-Conventional Yeast Yarrowia lipolytica. J. Microbiol. Methods 2017, 139, 161–164. [Google Scholar] [CrossRef]
- Vidal, L.; Lebrun, E.; Park, Y.K.; Mottet, G.; Nicaud, J.M. Bidirectional Hybrid Erythritol-Inducible Promoter for Synthetic Biology in Yarrowia lipolytica. Microb. Cell Fact. 2023, 22, 7. [Google Scholar] [CrossRef]
- Park, Y.-K.; Korpys, P.; Kubiak, M.; Celińska, E.; Soudier, P.; Trébulle, P.; Larroude, M.; Rossignol, T.; Nicaud, J. Engineering the Architecture of Erythritol-Inducible Promoters for Regulated and Enhanced Gene Expression in Yarrowia lipolytica. FEMS Yeast Res. 2019, 19, foy105. [Google Scholar] [CrossRef]
- Celińska, E.; Korpys-Woźniak, P.; Gorczyca, M.; Nicaud, J. Using Euf1 transcription factor as a titrator of erythritol-inducible promoters in Yarrowia lipolytica; insight into the structure, splicing, and regulation mechanism. FEMS Yeast Res. 2024, foae027. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Vandermies, M.; Soudier, P.; Telek, S.; Thomas, S.; Nicaud, J.M.; Fickers, P. Efficient Expression Vectors and Host Strain for the Production of Recombinant Proteins by Yarrowia lipolytica in Process Conditions. Microb. Cell Fact. 2019, 18, 167. [Google Scholar] [CrossRef] [PubMed]
- Trassaert, M.; Vandermies, M.; Carly, F.; Denies, O.; Thomas, S.; Fickers, P.; Nicaud, J.M. New Inducible Promoter for Gene Expression and Synthetic Biology in Yarrowia lipolytica. Microb. Cell Fact. 2017, 16, 141. [Google Scholar] [CrossRef] [PubMed]
- Korpys-Woźniak, P.; Kubiak, P.; Białas, W.; Celińska, E. Impact of Overproduced Heterologous Protein Characteristics on Physiological Response in Yarrowia lipolytica Steady-State-Maintained Continuous Cultures. Appl. Microbiol. Biotechnol. 2020, 104, 9785–9800. [Google Scholar] [CrossRef] [PubMed]
- Korpys-Woźniak, P.; Kubiak, P.; Celińska, E. Secretory Helpers for Enhanced Production of Heterologous Proteins in Yarrowia lipolytica. Biotechnol. Rep. 2021, 32, e00669. [Google Scholar] [CrossRef]
- Korpys-Woźniak, P.; Celińska, E. Global Transcriptome Profiling Reveals Genes Responding to Overproduction of a Small Secretory, a High Cysteine- and a High Glycosylation-Bearing Protein in Yarrowia lipolytica. Biotechnol. Rep. 2021, 31, e00646. [Google Scholar] [CrossRef]
- Oh, M.H.; Cheon, S.A.; Kang, H.A.; Kim, J.Y. Functional Characterization of the Unconventional Splicing of Yarrowia lipolytica HAC1 MRNA Induced by Unfolded Protein Response. Yeast 2010, 27, 443–452. [Google Scholar] [CrossRef]
- Guerfal, M.; Ryckaert, S.; Jacobs, P.P.; Ameloot, P.; Van Craenenbroeck, K.; Derycke, R.; Callewaert, N. The HAC1 Gene from Pichia Pastoris: Characterization and Effect of Its Overexpression on the Production of Secreted, Surface Displayed and Membrane Proteins. Microb. Cell Fact. 2010, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Korpys-Woźniak, P.; Celińska, E. Molecular Background of HAC1-Driven Improvement in the Secretion of Recombinant Protein in Yarrowia lipolytica Based on Comparative Transcriptomics. Biotechnol. Rep. 2023, 38, e00801. [Google Scholar] [CrossRef] [PubMed]
- Leplat, C.; Nicaud, J.-M.M.; Rossignol, T. Overexpression Screen Reveals Transcription Factors Involved in Lipid Accumulation in Yarrowia lipolytica. FEMS Yeast Res. 2018, 18, foy037. [Google Scholar] [CrossRef]
- Trébulle, P.; Nicaud, J.M.; Leplat, C.; Elati, M. Inference and Interrogation of a Coregulatory Network in the Context of Lipid Accumulation in Yarrowia lipolytica. NPJ Syst. Biol. Appl. 2017, 3, 21. [Google Scholar] [CrossRef]
- Gorczyca, M.; Nicaud, J.-M.; Celińska, E. Transcription Factors Enhancing Synthesis of Recombinant Proteins and Resistance to Stress in Yarrowia lipolytica. Appl. Microbiol. Biotechnol. 2023, 107, 4853–4871. [Google Scholar] [CrossRef] [PubMed]
- Gorczyca, M.; Białas, W.; Nicaud, J.-M.; Celińska, E. ‘Mother(Nature) Knows Best’—Hijacking Nature-Designed Transcriptional Programs for Enhancing Stress Resistance and Protein Production in Yarrowia lipolytica; Presentation of YaliFunTome Database. Microb. Cell Fact. 2024, 23, 26. [Google Scholar] [CrossRef]
- Ruchala, J.; Kurylenko, O.O.; Soontorngun, N.; Dmytruk, K.V.; Sibirny, A.A. Transcriptional Activator Cat8 Is Involved in Regulation of Xylose Alcoholic Fermentation in the Thermotolerant Yeast Ogataea (Hansenula) Polymorpha. Microb. Cell Fact. 2017, 16, 36. [Google Scholar] [CrossRef]
- Semkiv, M.V.; Ruchala, J.; Tsaruk, A.Y.; Zazulya, A.Z.; Vasylyshyn, R.V.; Dmytruk, O.V.; Zuo, M.X.; Kang, Y.; Dmytruk, K.V.; Sibirny, A.A. The Role of Hexose Transporter-like Sensor Hxs1 and Transcription Activator Involved in Carbohydrate Sensing Azf1 in Xylose and Glucose Fermentation in the Thermotolerant Yeast Ogataea Polymorpha. Microb. Cell Fact. 2022, 21, 162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, L.; Liang, S.; Zhang, P.; Kang, R.; Zhang, M.; Wang, M.; Chen, L.; Yuan, H.; Ding, S.; et al. FpDep1, a Component of Rpd3L Histone Deacetylase Complex, Is Important for Vegetative Development, ROS Accumulation, and Pathogenesis in Fusarium Pseudograminearum. Fungal Genet. Biol. 2020, 135, 103299. [Google Scholar] [CrossRef] [PubMed]
- Bonander, N.; Ferndahl, C.; Mostad, P.; Wilks, M.D.B.; Chang, C.; Showe, L.; Gustafsson, L.; Larsson, C.; Bill, R.M. Transcriptome Analysis of a Respiratory Saccharomycescerevisiae Strain Suggests the Expression of Its Phenotype Is Glucose Insensitive and Predominantly Controlled by Hap4, Cat8 and Mig1. BMC Genom. 2008, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Kricke, J.; Becher, D.; Lisowsky, T. Azf1p Is a Nuclear-Localized Zinc-Finger Protein That Is Preferentially Expressed under Non-Fermentative Growth Conditions in Saccharomyces cerevisiae. Curr. Genet. 1998, 34, 287–296. [Google Scholar] [CrossRef]
- Gorczyca, M.; Każmierczak, J.; Fickers, P.; Celińska, E. Synthesis of Secretory Proteins in Yarrowia lipolytica: Effect of Combined Stress Factors and Metabolic Load. Int. J. Mol. Sci. 2022, 23, 3602. [Google Scholar] [CrossRef]
- Gorczyca, M.; Kaźmierczak, J.; Steels, S.; Fickers, P.; Celińska, E. Impact of Oxygen Availability on Heterologous Geneexpression and Polypeptide Secretion Dynamics in Yarrowia lipolytica-based Protein Production Platforms. Yeast 2020, 37, 559–568. [Google Scholar] [CrossRef]
- Celińska, E.; Gorczyca, M. ‘Small Volume—Big Problem’: Culturing Yarrowia lipolytica in High-Throughput Micro-Formats. Microb. Cell Fact. 2024, 23, 184. [Google Scholar] [CrossRef] [PubMed]
- Doughty, T.; Kerkhoven, E. Extracting Novel Hypotheses and Findings from RNA-Seq Data. FEMS Yeast Res. 2020, 20, foaa007. [Google Scholar] [CrossRef]
- Li, J.; Singh, U.; Arendsee, Z.; Wurtele, E.S. Landscape of the Dark Transcriptome Revealed Through Re-Mining Massive RNA-Seq Data. Front. Genet. 2021, 12, 722981. [Google Scholar] [CrossRef] [PubMed]
- Sazegari, S.; Niazi, A.; Zinati, Z.; Eskandari, M.H. Mining Transcriptomic Data to Identify Saccharomyces cerevisiae Signatures Related to Improved and Repressed Ethanol Production under Fermentation. PLoS ONE 2022, 17, e0259476. [Google Scholar] [CrossRef]
- Abid, D.; Brent, M.R. NetProphet 3: A Machine Learning Framework for Transcription Factor Network Mapping and Multi-Omics Integration. Bioinformatics 2023, 39, btad038. [Google Scholar] [CrossRef]
- Sastry, A.V.; Poudel, S.; Rychel, K.; Yoo, R.; Lamoureux, C.R.; Chauhan, S.; Haiman, Z.B.; Al Bulushi, T.; Seif, Y.; Palsson, B.O. Mining All Publicly Available Expression Data to Compute Dynamic Microbial Transcriptional Regulatory Networks. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rychel, K.; Decker, K.; Sastry, A.V.; Phaneuf, P.V.; Poudel, S.; Palsson, B.O. IModulonDB: A Knowledgebase of Microbial Transcriptional Regulation Derived from Machine Learning. Nucleic Acids Res. 2021, 49, D112–D120. [Google Scholar] [CrossRef]
- Lee, S.-I.; Batzoglou, S. Application of Independent Component Analysis to Microarrays. Genome Biol. 2003, 4, R76. [Google Scholar] [CrossRef] [PubMed]
- Kerssemakers, A.; Krishnan, J.; Rychel, K.; Zielinski, D.; Palsson, B.; Sudarsan, S. Deciphering the TRN of Yarrowia lipolytica Using Machine Learning; Technical University of Denmark: Lyngby, Denmark, 2023. [Google Scholar]
- Kubiak-Szymendera, M.; Skupien-Rabian, B.; Jankowska, U.; Celińska, E. Hyperosmolarity Adversely Impacts Recombinant Protein Synthesis by Yarrowia lipolytica—Molecular Background Revealed by Quantitative Proteomics. Appl. Microbiol. Biotechnol. 2022, 106, 349–367. [Google Scholar] [CrossRef]
- Morales-Vargas, A.T.; Domínguez, A.; Ruiz-Herrera, J. Identification of Dimorphism-Involved Genes of Yarrowia lipolytica by Means of Microarray Analysis. Res. Microbiol. 2012, 163, 378–387. [Google Scholar] [CrossRef]
- Pomraning, K.R.; Bredeweg, E.L.; Baker, S.E. Regulation of Nitrogen Metabolism by GATA Zinc Finger Transcription Factors in Yarrowia lipolytica. mSphere 2017, 2, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Tsirigka, A.; Theodosiou, E.; Patsios, S.I.; Tsoureki, A.; Andreadelli, A.; Papa, E.; Aggeli, A.; Karabelas, A.J.; Makris, A.M. Novel Evolved Yarrowia lipolytica Strains for Enhanced Growth and Lipid Content under High Concentrations of Crude Glycerol. Microb. Cell Fact. 2023, 22, 62. [Google Scholar] [CrossRef]
- Wang, G.; Li, D.; Miao, Z.; Zhang, S.; Liang, W.; Liu, L. Comparative Transcriptome Analysis Reveals Multiple Functions for Mhy1p in Lipid Biosynthesis in the Oleaginous Yeast Yarrowia lipolytica. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Konzock, O.; Norbeck, J. Deletion of MHY1 Abolishes Hyphae Formation in Yarrowia lipolytica without Negative Effects on Stress Tolerance. PLoS ONE 2020, 15, e0231161. [Google Scholar] [CrossRef] [PubMed]
- Shu, T.; He, X.-Y.; Chen, J.-W.; Mao, Y.-S.; Gao, X.-D. The PH-Responsive Transcription Factors YlRim101 and Mhy1 Regulate Alkaline PH-Induced Filamentation in the Dimorphic Yeast Yarrowia lipolytica. mSphere 2021, 6, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vazquez, A.; Gonzalez-Hernandez, A.; Domínguez, Á.; Rachubinski, R.; Riquelme, M.; Cuellar-Mata, P.; Guzman, J.C.T. Identification of the Transcription Factor Znc1p, Which Regulates the Yeast-to-Hypha Transition in the Dimorphic Yeast Yarrowia lipolytica. PLoS ONE 2013, 8, e66790. [Google Scholar] [CrossRef]
- Mittal, N.; Guimaraes, J.C.; Gross, T.; Schmidt, A.; Vina-Vilaseca, A.; Nedialkova, D.D.; Aeschimann, F.; Leidel, S.A.; Spang, A.; Zavolan, M. The Gcn4 Transcription Factor Reduces Protein Synthesis Capacity and Extends Yeast Lifespan. Nat. Commun. 2017, 8, 457. [Google Scholar] [CrossRef]
- Zhang, L.; Hach, A. Molecular Mechanism of Heme Signaling in Yeast: The Transcriptional Activator Hap1 Serves as the Key Mediator. Cell Mol. Life Sci. 1999, 56, 415–426. [Google Scholar] [CrossRef]
- Rzechonek, D.A.; Szczepańczyk, M.; Borodina, I.; Neuvéglise, C.; Mirończuk, A.M. Transcriptome Analysis Reveals Multiple Targets of Erythritol-Related Transcription Factor EUF1 in Unconventional Yeast Yarrowia lipolytica. Microb. Cell Fact. 2024, 23, 77. [Google Scholar] [CrossRef]
- Rzechonek, D.A.; Neuvéglise, C.; Devillers, H.; Rymowicz, W.; Mirończuk, A.M. EUF1-A Newly Identified Gene Involved in Erythritol Utilization in Yarrowia lipolytica. Sci. Rep. 2017, 7, 12507. [Google Scholar] [CrossRef]
- Hirakawa, K.; Kobayashi, S.; Inoue, T.; Endoh-Yamagami, S.; Fukuda, R.; Ohta, A. Yas3p, an Opi1 Family Transcription Factor, Regulates Cytochrome P450 Expression in Response to n-Alkanes in Yarrowia lipolytica. J. Biol. Chem. 2009, 284, 7126–7137. [Google Scholar] [CrossRef]
- Pomraning, K.R.; Bredeweg, E.L.; Kerkhoven, E.J.; Barry, K.; Haridas, S.; Hundley, H.; LaButti, K.; Lipzen, A.; Yan, M.; Magnuson, J.K.; et al. Regulation of Yeast-to-Hyphae Transition in Yarrowia lipolytica. mSphere 2018, 3, e00541-18. [Google Scholar] [CrossRef]
- Mao, Y.S.; Chen, J.W.; Wang, Z.H.; Xu, M.Y.; Gao, X.D. Roles of the Transcriptional Regulators Fts1, YlNrg1, YlTup1, and YlSsn6 in the Repression of the Yeast-to-Filament Transition in the Dimorphic Yeast Yarrowia lipolytica. Mol. Microbiol. 2023, 119, 126–142. [Google Scholar] [CrossRef]
- Celińska, E.; Borkowska, M.; Białas, W.; Kubiak, M.; Korpys, P.; Archacka, M.; Ledesma-Amaro, R.; Nicaud, J.M. Genetic Engineering of Ehrlich Pathway Modulates Production of Higher Alcohols in Engineered Yarrowia lipolytica. FEMS Yeast Res. 2019, 19, foy122. [Google Scholar] [CrossRef]
- García, S.; Prado, M.; Dégano, R.; Domínguez, A. A Copper-Responsive Transcription Factor, CRF1, Mediates Copper and Cadmium Resistance in Yarrowia lipolytica. J. Biol. Chem. 2002, 277, 37359–37368. [Google Scholar] [CrossRef]
- Wang, Z.-P.; Xu, H.-M.; Wang, G.-Y.; Chi, Z.; Chi, Z.-M. Disruption of the MIG1 Gene Enhances Lipid Biosynthesis in the Oleaginous Yeast Yarrowia lipolytica ACA-DC 50109. Biochim. Biophys. Acta 2013, 1831, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Torres-Guzma’n, J.C.; Guzma´n, G.; Domi´nguez, A.; Domi´nguez, D. HOY1, a Homeo Gene Required for Hyphal Formation in Yarrowia lipolytica. Mol. Cell. Biol. 1997, 17, 6283–6293. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-W.; Mao, Y.-S.; Yan, L.-Q.; Gao, X.-D. The Zinc Finger Transcription Factor Fts2 Represses the Yeast-to-Filament Transition in the Dimorphic Yeast Yarrowia lipolytica. mSphere 2022, 7, e0045022. [Google Scholar] [CrossRef] [PubMed]
- González-Lozano, K.J.; Aréchiga-Carvajal, E.T.; Jiménez-Salas, Z.; Valdez-Rodríguez, D.M.; León-Ramírez, C.G.; Ruiz-Herrera, J.; Adame-Rodríguez, J.M.; López-Cabanillas-Lomelí, M.; Campos-Góngora, E. Identification and Characterization of Dmct: A Cation Transporter in Yarrowia lipolytica Involved in Metal Tolerance. J. Fungi 2023, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Ryu, S.; Trinh, C.T. Exceptional Solvent Tolerance in Yarrowia lipolytica Is Enhanced by Sterols. Metab. Eng. 2019, 54, 83–95. [Google Scholar] [CrossRef]
- Poopanitpan, N.; Kobayashi, S.; Fukuda, R.; Horiuchi, H.; Ohta, A. An Ortholog of FarA of Aspergillus Nidulans Is Implicated in the Transcriptional Activation of Genes Involved in Fatty Acid Utilization in the Yeast Yarrowia lipolytica. Biochem. Biophys. Res. Commun. 2010, 402, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Endoh-Yamagami, S.; Hirakawa, K.; Morioka, D.; Fukuda, R.; Ohta, A. Basic Helix-Loop-Helix Transcription Factor Heterocomplex of Yas1p and Yas2p Regulates Cytochrome P450 Expression in Response to Alkanes in the Yeast Yarrowia lipolytica. Eukaryot. Cell 2007, 6, 734–743. [Google Scholar] [CrossRef]
- Hurtado, C.A.R.; Rachubinski, R.A. Mhy1 Encodes a C2h2-Type Zinc Finger Protein That Promotes Dimorphic Transition in the Yeast Yarrowia lipolytica. J. Bacteriol. 1999, 181, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, C.A.R.; Rachubinski, R.A. YlBMH1 Encodes a 14-3-3 Protein That Promotes Filamentous Growth in the Dimorphic Yeast Yarrowia lipolytica. Microbiology 2002, 148, 3725–3735. [Google Scholar] [CrossRef]
- Celińska, E.; Olkowicz, M.; Grajek, W. L-Phenylalanine Catabolism and 2-Phenylethano Synthesis in Yarrowia lipolytica-Mapping Molecular Identities through Whole-Proteome Quantitative Mass Spectrometry Analysis. FEMS Yeast Res. 2015, 15, fov041. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P. Protocol Update for Large-Scale Genome and Gene Function Analysis with PANTHER Classification System (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER Version 16: A Revised Family Classification, Tree-Based Classification Tool, Enhancer Regions and Extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Mogi, R.; Watanabe, J. Identification of SFL1 as a Positive Regulator for Flor Formation in Zygosaccharomyces Rouxii. Biosci. Biotechnol. Biochem. 2020, 84, 1291–1298. [Google Scholar] [CrossRef]
- Ansanay Galeote, V.; Alexandre, H.; Bach, B.; Delobel, P.; Dequin, S.; Blondin, B. Sfl1p Acts as an Activator of the HSP30 Gene in Saccharomyces cerevisiae. Curr. Genet. 2007, 52, 55–63. [Google Scholar] [CrossRef]
- Atsushi, F.; Yoshiko, K.; Satoru, K.; Yoshio, M.; Shinichi, M.; Harumi, K. Domains of the SFL1 Protein of Yeasts Are Homologous to Myc Oncoproteins or Yeast Heat-Shock Transcription Factor. Gene 1989, 85, 321–328. [Google Scholar] [CrossRef]
- De Vit, M.J.; Waddle, J.A.; Johnston, M. Regulated Nuclear Translocation of the Mig1 Glucose Repressor. Mol. Biol. Cell 1997, 8, 1603–1618. [Google Scholar] [CrossRef]
- Treitel, M.A.; Carlson, M. Repression by SSN6-TUP1 Is Directed by MIG1, a Repressor/Activator Protein (Transcription/Yeast/Zinc-Finger Protein/Glucose Repression). Proc. Natl. Acad. Sci. USA 1995, 92, 3132–3136. [Google Scholar] [CrossRef]
- Martínez, J.L.; Meza, E.; Petranovic, D.; Nielsen, J. The Impact of Respiration and Oxidative Stress Response on Recombinant α-Amylase Production by Saccharomyces cerevisiae. Metab. Eng. Commun. 2016, 3, 205–210. [Google Scholar] [CrossRef]
- Rep, M.; Proft, M.; Remize, F.; Tamás, M.; Serrano, R.; Thevelein, J.M.; Hohmann, S. The Saccharomyces cerevisiae Sko1p Transcription Factor Mediates HOG Pathway-Dependent Osmotic Regulation of a Set of Genes Encoding Enzymes Implicated in Protection from Oxidative Damage. Mol. Microbiol. 2001, 40, 1067–1083. [Google Scholar] [CrossRef]
- Karlgren, S.; Pettersson, N.; Nordlander, B.; Mathai, J.C.; Brodsky, J.L.; Zeidel, M.L.; Bill, R.M.; Hohmann, S. Conditional Osmotic Stress in Yeast: A System to Study Transport through Aquaglyceroporins and Osmostress Signaling. J. Biol. Chem. 2004, 280, 7186–7193. [Google Scholar] [CrossRef]
- Callewaer, N.; Vervecken, W.; De Pourq, K.; Geysens, S.; Guerfal, M. Yarrowia lipolytica and Pichia Pastoris Hac1 Nucleic Acids. U.S. Patent US8026083B2, 27 September 2011. [Google Scholar]
- Hooks, K.B.; Griffiths-Jones, S. Conserved RNA Structures in the Non-Canonical Hac1/Xbp1 Intron. RNA Biol. 2011, 8, 552–556. [Google Scholar] [CrossRef]
- Whyteside, G.; Nor, R.M.; Alcocer, M.J.C.; Archer, D.B. Activation of the Unfolded Protein Response in Pichia Pastoris Requires Splicing of a HAC1 MRNA Intron and Retention of the C-Terminal Tail of Hac1p. FEBS Lett. 2011, 585, 1037–1041. [Google Scholar] [CrossRef]
- Graf, A.; Dragosits, M.; Gasser, B.; Mattanovich, D. Yeast Systems Biotechnology for the Production of Heterologous Proteins. FEMS Yeast Res. 2009, 9, 335–348. [Google Scholar] [CrossRef]
- Zahrl, R.J.; Gasser, B.; Mattanovich, D.; Ferrer, P. Detection and Elimination of Cellular Bottlenecks in Protein-Producing Yeasts. Methods Mol. Biol. 2019, 1923, 75–95. [Google Scholar] [CrossRef]
- Gasser, B.; Sauer, M.; Maurer, M.; Stadlmayr, G.; Mattanovich, D. Transcriptomics-Based Identification of Novel Factors Enhancing Heterologous Protein Secretion in Yeasts. Appl. Environ. Microbiol. 2007, 73, 6499–6507. [Google Scholar] [CrossRef]
- Zahrl, R.J.; Prielhofer, R.; Burgard, J.; Mattanovich, D.; Gasser, B. Synthetic Activation of Yeast Stress Response Improves Secretion of Recombinant Proteins. N. Biotechnol. 2023, 73, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Gasser, B.; Maurer, M.; Rautio, J.; Sauer, M.; Bhattacharyya, A.; Saloheimo, M.; Penttilä, M.; Mattanovich, D. Monitoring of Transcriptional Regulation in Pichia Pastoris under Protein Production Conditions. BMC Genom. 2007, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Zahrl, R.J.; Peña, D.A.; Mattanovich, D.; Gasser, B. Systems Biotechnology for Protein Production in Pichia Pastoris. FEMS Yeast Res. 2017, 17, fox068. [Google Scholar] [CrossRef] [PubMed]
- Lamping, E.; Lückl, J.; Paltauf, F.; Henry, S.A.; Kohlwein, S.D. Isolation and Characterization of a Mutant of Saccharomyces cerevisiae with Pleiotropic Deficiencies in Transcriptional Activation and Repression. Genetics 1994, 137, 55–65. [Google Scholar] [CrossRef]
- Shimanuki, M.; Uehara, L.; Pluskal, T.; Yoshida, T.; Kokubu, A.; Kawasaki, Y.; Yanagida, M. Klf1, a C2H2 Zinc Finger-Transcription Factor, Is Required for Cell Wall Maintenance during Long-Term Quiescence in Differentiated G0 Phase. PLoS ONE 2013, 8, e78545. [Google Scholar] [CrossRef]
- Herholz, M.; Cepeda, E.; Baumann, L.; Kukat, A.; Hermeling, J.; Maciej, S.; Szczepanowska, K.; Pavlenko, V.; Frommolt, P.; Trifunovic, A. KLF-1 Orchestrates a Xenobiotic Detoxification Program Essential for Longevity of Mitochondrial Mutants. Nat. Commun. 2019, 10, 3323. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Österlund, T.; Liu, Z.; Petranovic, D.; Nielsen, J.; Osterlund, T.; Liu, Z.; Petranovic, D.; Nielsen, J. Heat Shock Response Improves Heterologous Protein Secretion in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2013, 97, 3559–3568. [Google Scholar] [CrossRef]
- Hou, J.; Tyo, K.E.J.; Liu, Z.; Petranovic, D.; Nielsen, J. Metabolic Engineering of Recombinant Protein Secretion by Saccharomyces cerevisiae. FEMS Yeast Res. 2012, 12, 491–510. [Google Scholar] [CrossRef]
- Hou, J.; Tang, H.; Liu, Z.; Österlund, T.; Nielsen, J.; Petranovic, D. Management of the Endoplasmic Reticulum Stress by Activation of the Heat Shock Response in Yeast. FEMS Yeast Res. 2014, 14, 481–494. [Google Scholar] [CrossRef]
- Leplat, C.; Nicaud, J.M.; Rossignol, T. High-Throughput Transformation Method for Yarrowia lipolytica Mutant Library Screening. FEMS Yeast Res. 2015, 15, fov052. [Google Scholar] [CrossRef]
- Celińska, E.; Ledesma-Amaro, R.; Larroude, M.; Rossignol, T.; Pauthenier, C.; Nicaud, J.M. Golden Gate Assembly System Dedicated to Complex Pathway Manipulation in Yarrowia lipolytica. Microb. Biotechnol. 2017, 10, 450–455. [Google Scholar] [CrossRef]
- Korpys-Woźniak, P.; Kubiak, M.; Borkowska, M.; Celińska, E. Construction and Assembly of Standardized Biobricks for Synthetic Pathways Engineering in Yeasts. In Synthetic Biology of Yeasts: Tools and Applications; Darvishi Harzevili, F., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 27–62. ISBN 978-3-030-89680-5. [Google Scholar]
- Celińska, E.; Borkowska, M.; Białas, W.; Korpys, P.; Nicaud, J.M. Robust Signal Peptides for Protein Secretion in Yarrowia lipolytica: Identification and Characterization of Novel Secretory Tags. Appl. Microbiol. Biotechnol. 2018, 102, 5221–5233. [Google Scholar] [CrossRef] [PubMed]
- Borkowska, M.; Białas, W.; Kubiak, M.; Celińska, E. Rapid Micro-Assays for Amylolytic Activities Determination: Customization and Validation of the Tests. Appl. Microbiol. Biotechnol. 2019, 103, 2367–2379. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; ISBN 978-0879695-76-7. [Google Scholar]
- Larroude, M.; Park, Y.K.; Soudier, P.; Kubiak, M.; Nicaud, J.M.; Rossignol, T. A Modular Golden Gate Toolkit for Yarrowia lipolytica Synthetic Biology. Microb. Biotechnol. 2019, 12, 1249–1259. [Google Scholar] [CrossRef]
- Larroude, M.; Trabelsi, H.; Nicaud, J.M.; Rossignol, T. A Set of Yarrowia lipolytica CRISPR/Cas9 Vectors for Exploiting Wild-Type Strain Diversity. Biotechnol. Lett. 2020, 42, 773–785. [Google Scholar] [CrossRef]
- Chen, D.C.; Beckerich, J.M.; Gaillardin, C. One-Step Transformation of the Dimorphic Yeast Yarrowia lipolytica. Appl. Microbiol. Biotechnol. 1997, 48, 232–235. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yali Signature | Assigned Name | Putative/Known Function | Reference |
|---|---|---|---|
| B12716 | HAC1 | Transcriptional activator of genes involved in ER-based Unfolded Protein Response (UPR). | [16,19] |
| E31757 | BRG1 | Biofilm regulator 1. | [20] |
| D14520 | SKN7 | TF involved in the activation of osmotic and oxidative stress. | [22,40] |
| D07744 | YAP-like | TF involved in pH-dependent dimorphic transition and maintaining redox balance. | [22,41] |
| D20482 | GZF1 | Inducer of the nitrogen catabolite repression (NCR) genes. | [23,42] |
| E13948 | HSF1 | Heat shock transcription factor. | [22,23] |
| D05041 | KLF1 | Krueppel-like factor 15, regulates the expression of genes for gluconeogenic and amino acid-degrading enzymes. | [23] |
| A16841 | AZF1 | Asparagine-rich Zinc Finger protein, regulates carbon metabolism in yeast and cell wall organization. | [20,23] |
| F05896 | DEP1 | Part of the Rpd3C(L) histone deacetylase complex (HDAC). Transcriptional modulator involved in regulation of structural phospholipid biosynthesis genes. | [20,23] |
| C19151 | CAT8 | CATabolite repression TF 8. Inducer of gluconeogesis, maintains energy homeostasis, presumed to regulate formate dehydrogenases expression. | [39] |
| F21923 | ADR1 | Alcohol Dehydrogenase II synthesis Regulator, inducer of genes involved in alternative carbon utilization upon glucose starvation. | [11,39] |
| D20460 | LAC9 | LACtose regulatory protein, controls induction of the lactose-galactose regulation. | [23] |
| B14443 | JMC2 | JmjC domain family histone demethylase, promotes global demethylation of H3K4. | [23,43] |
| D02783 | DAL81 | Positive regulator of genes in multiple nitrogen degradation pathways, involved in nitrogen catabolite activation of transcription from RNA polymerase II promoter. | [23] |
| B19602 | MGF2 | Mycelial growth factor 2. TF of known roles in the dimorphic transition. | [16,41] |
| B21582 | MHY1 | Mns2/Mns4-like protein, a key regulator of yeast-to hypha dimorphic transition but not stress resistance, regulates both alkaline-pH and glucose-induced filamentation. | [44,45,46] |
| D01573 | MGF1 | Mycelial growth factor. Potential driver of the transition between morphological phases. | [16] |
| B05038 | ZNC1 | Zinc finger transcriptional factor, regulates the yeast-to-hypha transition in the dimorphic yeast. | [47] |
| F17886 | GZF2 | GATA—binding zinc finger transcription factor 2, inducer of NCR, essential for growth on simple nitrogen sources. | [42] |
| C22682 | GZF3 | GATA-zinc finger transcription factor 3, repressor of NCR. | [20,42] |
| E05555 | GZF4 | GATA-zinc finger transcription factor 4. Putative: inducer of NCR. | [42] |
| E16577 | GZF5 | Non-genuine GATA-zinc finger transcription factor. | [20,42] |
| E03410 | ERT1-2 | Positive regulator of gluconeogenesis. | [20] |
| E27742 | GCN4 | General Control Non-derepressible 4 TF. Key transcriptional activator of amino acid biosynthesis genes. | [48] |
| F17424 | HAP1 | TF responsible for oxygen sensing and signaling. | [49] |
| D04785 | SFL1 | Repressor of filamentous growth and flocculation. | |
| F01562 | EUF1 | Transcription factor mediating expression of erythritol synthesis genes. | [11,50,51] |
| C16863 | SKO1 | ATF/CREB family transcription factor, repressor that mediates HOG pathway-dependent regulation of osmotic stress response, involved in protection from oxidative damage. | [40] |
| C14784 | YAS3 | Transcriptional repressor of ALK genes, de-repressed on alkanes. | [52] |
| C13750 | MSN4 | General stress response, regulates tolerance to acid-induced stress. Regulates genes involved in the antioxidant cellular response. | [53] |
| B13640 | RIM101 | pH-response transcription factor, regulator of alkaline-induced filamentation | [46] |
| C12364 | NRG1 | NRG1 repressor of erythritol utilization genes, plays a minor role in repression of filamentation. | [50,54] |
| D23749 | ZAP1 | ZAP1 involved in zinc ion homeostasis by zinc-responsive transcriptional regulation. | [20] |
| E30789 | Putativea: transcriptional regulator of form adherence 5. | ||
| C07821 | Putativea: glucose transport transcription regulator RGT1-related. | ||
| C18645 | ARO80 | Transcription activator required for the expression of genes involved in the catabolism of aromatic amino acids. | [55] |
| B08206 | CRF1 | Copper resistance protein transcriptional regulator. | [56] |
| D14872 | Putative a: transcriptional regulatory protein STB4 | ||
| E18304 | ERT1-1 | Transcription activator of gluconeogenesis. | [20] |
| E07942 | MIG1 | Controls genes involved in beta-oxidation, involved in carbon catabolite repression. | [57] |
| A18469 | HOY1 | Homeobox protein, a positive regulator of hyphae formation. | [20,58] |
| B06853 | PUT3 | PUT3 proline utilization trans-activator. | |
| B09713 | PPR1 | PPR1 pyrimidine pathway regulatory protein, de novo biosynthesis. | [54,59] |
| D13904 | LEU3 | Leucine-responsive transcription regulator, regulates genes involved in branched-chain amino acid biosynthesis and ammonia assimilation. | |
| D23045 | AHR1 | AHR1 adhesion and hyphal regulator 1. | |
| E10681 | WAR1 | Weak Acid Resistance transcription factor. | |
| E20449 | YOX1 | Homeobox protein YOX1. Transcriptional repressor of ECB-dependent genes (early cell box) to the G1/M phase. | |
| F03157 | MET32 | MET32 auxiliary transcriptional regulator of sulfonate and sulfur amino acid metabolism, methionine biosynthesis, and sulfate assimilation. | |
| F05104 | TFIIIA | PZF1 general transcription factor IIIA. | |
| F09361 | U4/U6.U5 component | U4/U6.U5 tri-snRNP, spliceosomal complex—may play a role in mRNA splicing. | |
| F09493 | SAP61 | Pre-mRNA-splicing factor sap61, involved in mRNA splicing, associates with cdc5 and the other cwf proteins as part of the spliceosome. | |
| F16599 | STB4 | Putative transcription factor STB4—Sin Three Binding protein, involved in the transcription of transmembrane transporters. | [60] |
| F18788 | RLM1 | May function as a TF downstream of MPK1, at least some RML1 target genes are involved in cell wall biosynthesis. | |
| E31845 | PRZ1 | Involved in the regulation of calcium ion homeostasis. | |
| B15818 | SterTF | Sterol transcription factor, regulating sterol biogenesis. | [61] |
| D05005 | SEF1 | Zn2-Cys6 transcription factor; regulates iron uptake. | |
| D12628 | POR1 | Primary oleate regulator 1—transcriptional activator regulating genes involved in fatty acid utilization. | [62] |
| C02387 | YAS1 | Transcription factor essential for cytochrome p450 induction in response to alcanes, heteromeric Yas1p/Yas2p complex transcription factor. | [63] |
| B08734 | REI1 | Cytoplasmic pre-60S factor REI1 involved in maturation of the ribosomal 60S subunit. | |
| E01606 | OAF3 | Oleate activated transcription factor 3, transcriptional inhibitor with a significantly increased number of target genes in response to oleate. | |
| C06842 | MCM1 | MCM1 transcription factor involved in biofilm formation, cell adhesion, and hyphal growth. | |
| A19778 | MBP1 | MluI-box Binding Protein, involved in regulation of cell cycle progression from G1 to S phase. | [39,53] |
| D15334 | CPH2 | Transcription factor that positively controls filamentous growth. | |
| D01463 | CRZ1 | CRZ1 transcription regulator involved in the regulation of calcium ion homeostasis. | |
| D13068 | BUD20 | BUD20 bud site selection protein 20—positioning the proximal bud pole signal; protein required for ribosome assembly. | |
| B13354 | AP-4 | Transcription factor that activates viral and cellular genes. | |
| F25861 | RPN4 | RPN4 transcription factor regulating proteasomal genes. | |
| D24167 | CBF1 | CBF1 centromere binding factor 1, required for chromosome stability and chromosomal segregation. | |
| C12639 | SWI6 | SWI6 part of a complex involved in cell-cycle-dependent transcription. SWI4 and SWI6 are required for formation of the cell-cycle box factor-DNA complex | |
| E25960 | SWI1 | SWI/SNF chromatin-remodeling complex subunit SWI1. | |
| F13321 | OAF1 | Oleate activated transcription factor 1, activates transcription of genes involved in fatty acid beta-oxidation. | |
| F11487 | STP1 | Nutrient- and stress-responsive activator of ribosome biogenesis genes. | |
| B05478 | STP3 | Possibly involved in pre-tRNA splicing and in uptake of branched-chain amino acids. | [59] |
| E15510 | Putative a: Homeobox protein YOX1. | ||
| C22990 | Putative a: ASG1 general activator of stress genes. | ||
| D04466 | Putative a: Regulatory protein cys-3. Turns on the expression of structural genes which encode sulfur-catabolic enzymes. | ||
| D09647 | Putative a: Arginine metabolism regulation protein II. With ARG80, ARG82 and MCM1, coordinates the expression of arginine anabolic and catabolic genes in response to arginine. | ||
| D10681 | Putative a: Adhesion and hyphal regulator 1. | ||
| D18678 | Putative a: Respiration factor 2. Transcription factor that regulates expression genes required for glycerol-based growth and respiration. | ||
| E17721 | Putative a: Phosphatidylinositol N-acetylglucosaminyltransferase subunit P. | ||
| E31669 | Putative a: Metal-binding activator 1. Copper ion-sensing transcription factor, promotes filamentous and invasive growth. | ||
| F05126 | Putative a: Phosphorus acquisition-controlling protein. | ||
| F17468 | Putative a: Multidrug resistance regulator 1. Acts as the central regulator of the MDR1 efflux pump. | ||
| E11693 | Putative a: TF required for repression of genes during iron starvation. Represses iron-dependent and mitochondrial-localized activities including respiration, TCA cycle, amino acid metabolism, iron-sulfur-cluster and heme biosynthesis. | ||
| E10131 | Putative a: Transcription elongation factor 1. Implicated in the maintenance of proper chromatin structure in actively transcribed regions. | ||
| D09757 | Putative a: AP-1-like transcription factor. | ||
| F05346 | Putative a: Binds a palindromic promoter element essential for induction of fungal cutinase gene. | ||
| C11858 | Putative a: DNA-binding transcription factor Moc3. | ||
| B15312 | Putative a: May act as a co-chaperone for HSP70. | ||
| C18667 | Putative a: Transcriptional activator of the arabinanolytic system. | ||
| E16973 | Putative a: Metallothionein expression activator. | ||
| F11979 | Putative a: Retrograde regulation protein 1. | ||
| C19063 | Putative a: Transcription factor that mediates stress and developmental response. | ||
| F18326, E05577, B08360, C20977, C13794 | Putative a: sterol uptake control. | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorczyca, M.; Korpys-Woźniak, P.; Celińska, E. An Interplay between Transcription Factors and Recombinant Protein Synthesis in Yarrowia lipolytica at Transcriptional and Functional Levels—The Global View. Int. J. Mol. Sci. 2024, 25, 9450. https://doi.org/10.3390/ijms25179450
Gorczyca M, Korpys-Woźniak P, Celińska E. An Interplay between Transcription Factors and Recombinant Protein Synthesis in Yarrowia lipolytica at Transcriptional and Functional Levels—The Global View. International Journal of Molecular Sciences. 2024; 25(17):9450. https://doi.org/10.3390/ijms25179450
Chicago/Turabian StyleGorczyca, Maria, Paulina Korpys-Woźniak, and Ewelina Celińska. 2024. "An Interplay between Transcription Factors and Recombinant Protein Synthesis in Yarrowia lipolytica at Transcriptional and Functional Levels—The Global View" International Journal of Molecular Sciences 25, no. 17: 9450. https://doi.org/10.3390/ijms25179450