
CEP-1347 Boosts Chk2-Mediated p53 Activation by Ionizing Radiation to Inhibit the Growth of Malignant Brain Tumor Cells


 , ,
, ,
Abstract
:1. Introduction
2. Results
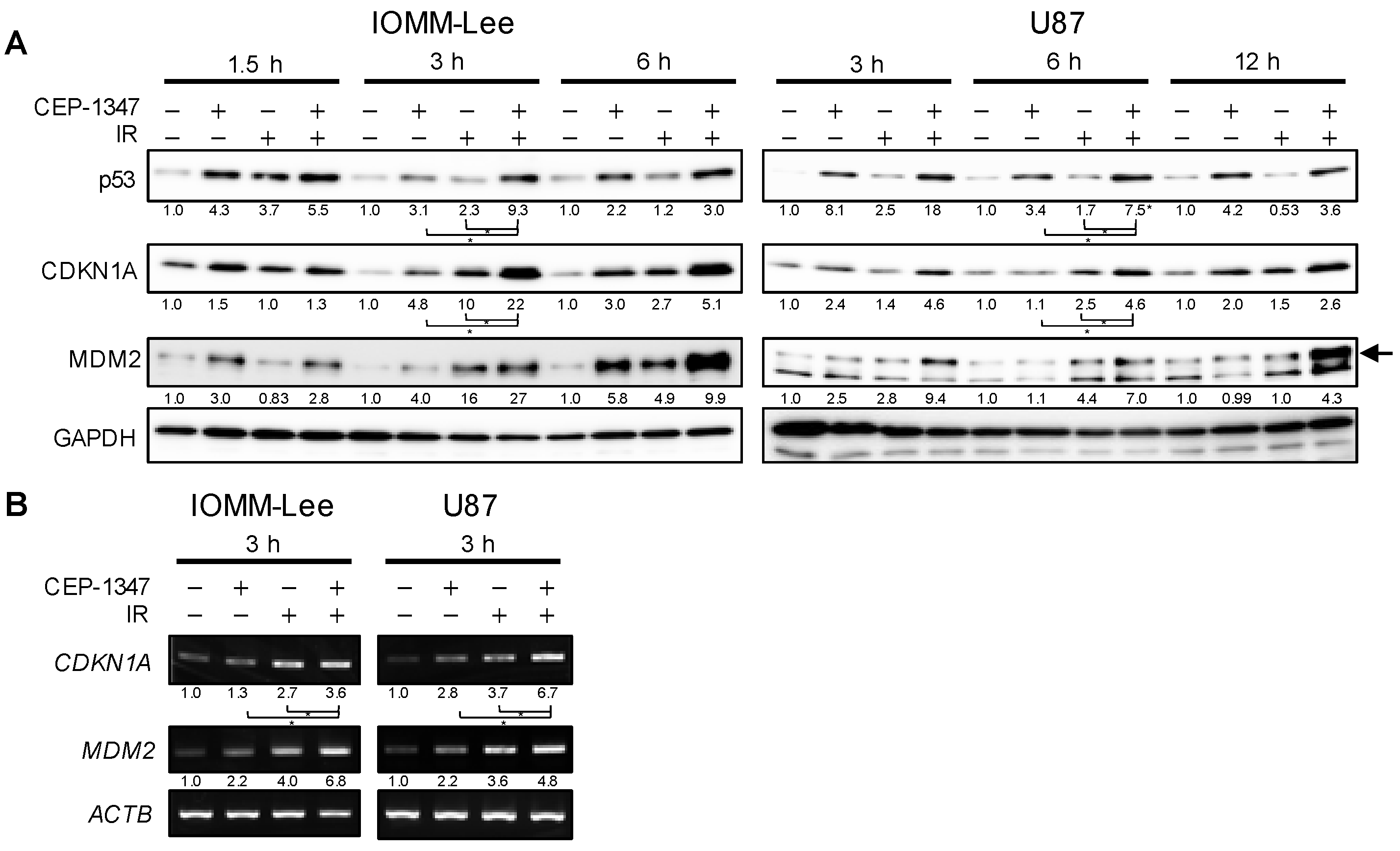
2.1. CEP-1347 Enhances Growth Inhibitory Effects of IR in Malignant Brain Tumor Cells and Amplifies Activation of the p53 Pathway by IR
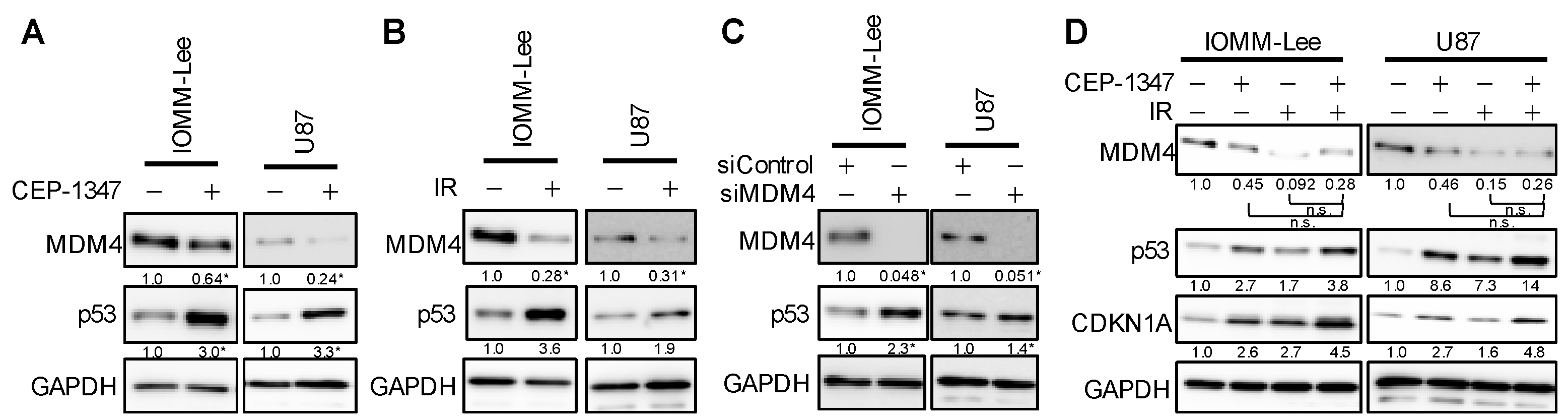
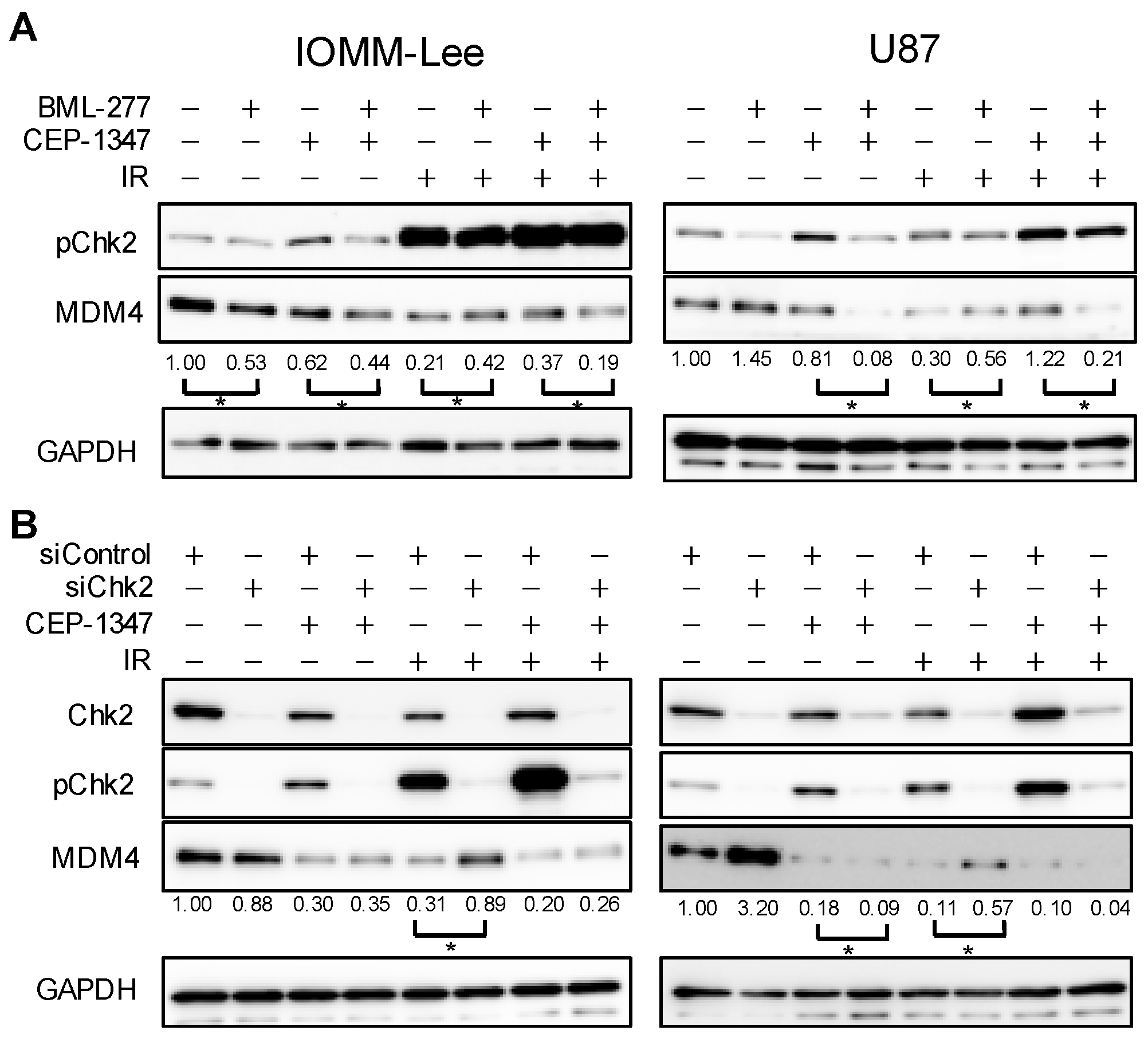
2.2. The Combination of CEP-1347 and IR Does Not Result in the Further Inhibition of MDM4 Expression
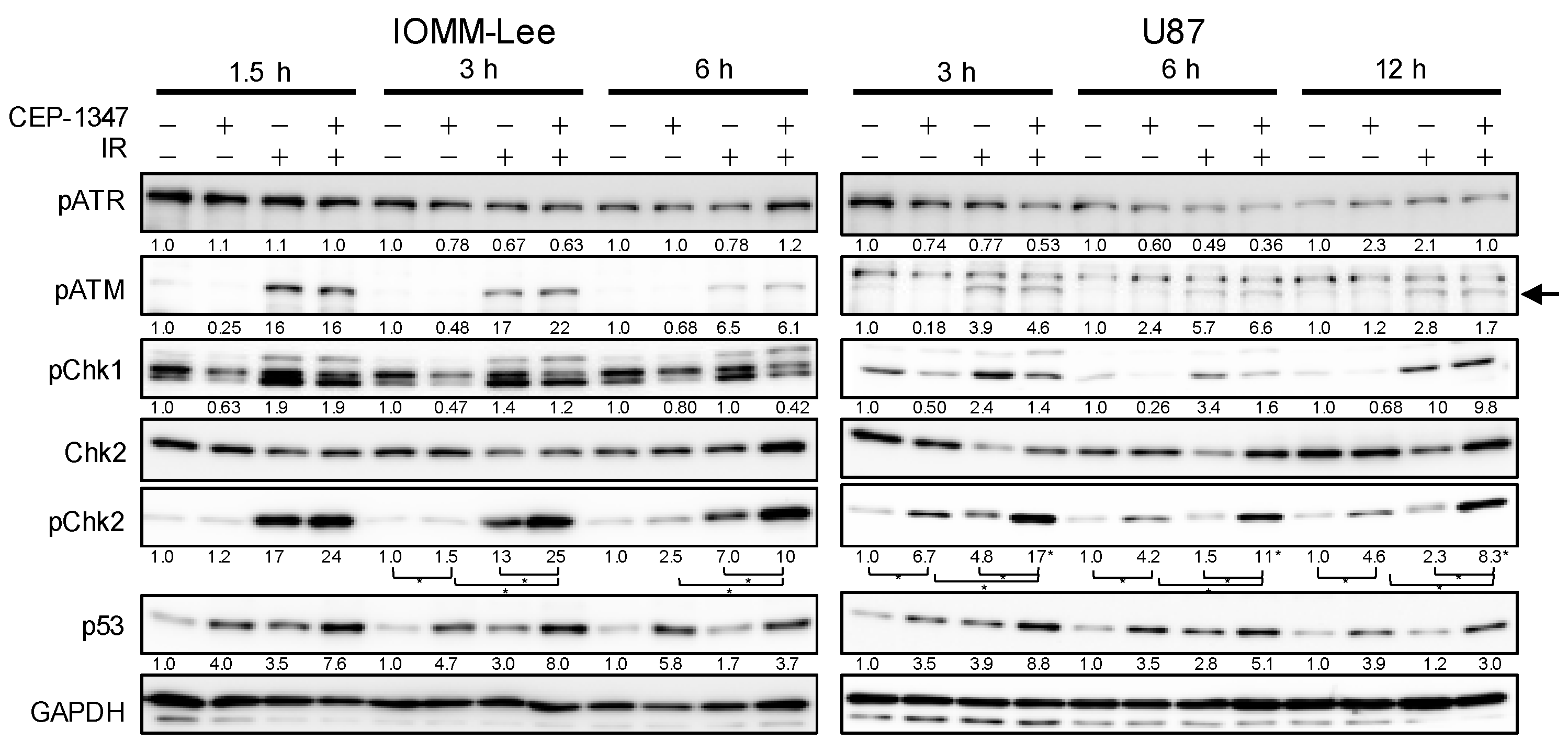
2.3. CEP-1347 Enhances IR-Mediated Increases in Chk2 Phosphorylation in an ATM-Independent Manner
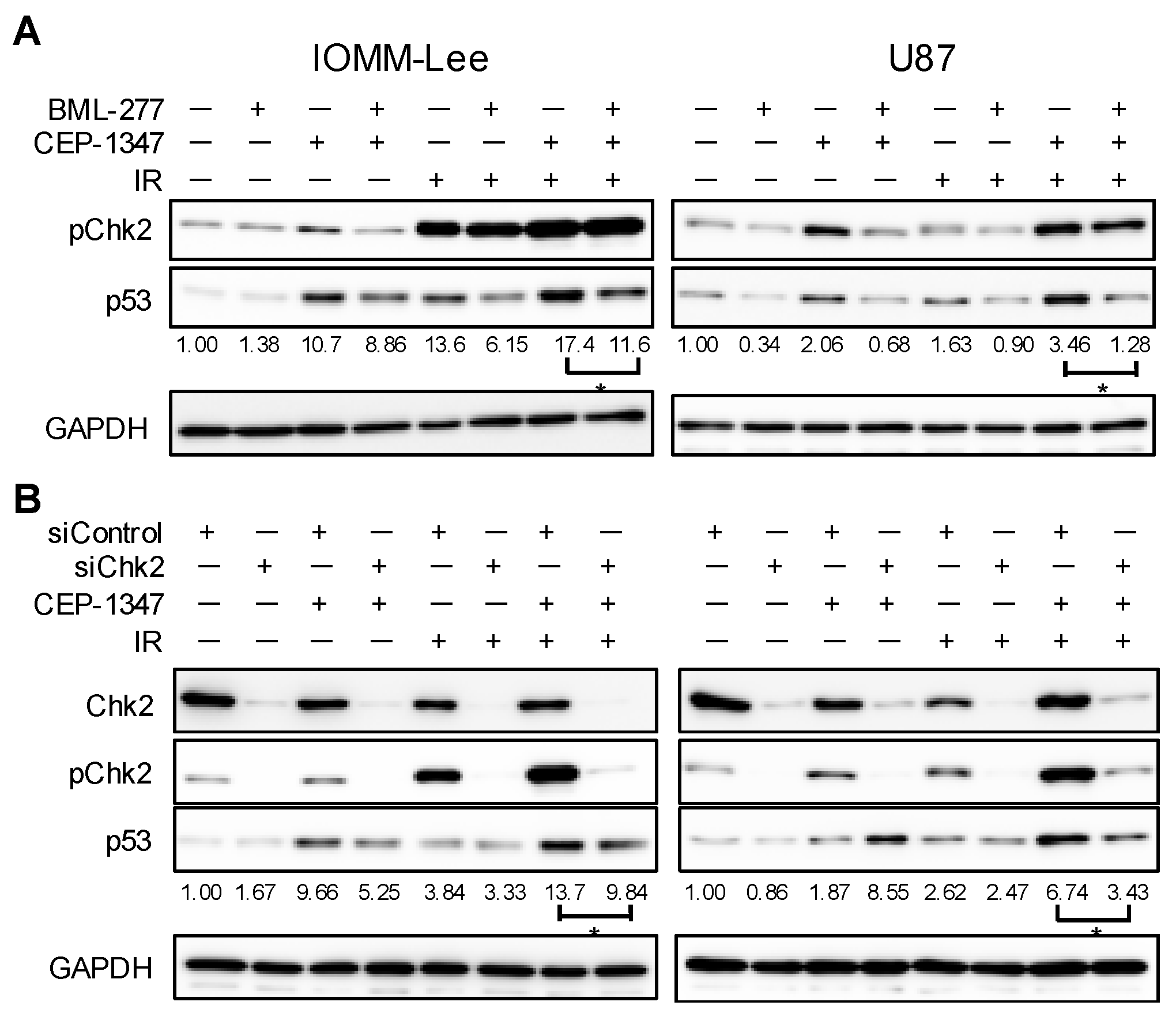
2.4. Chk2 Activity Is Necessary for CEP-1347 Activation of p53 in Combination with IR
2.5. MDM4 Expression Is Down-Regulated by IR and CEP-1347 in Chk2-Dependent and Independent Manners, Respectively
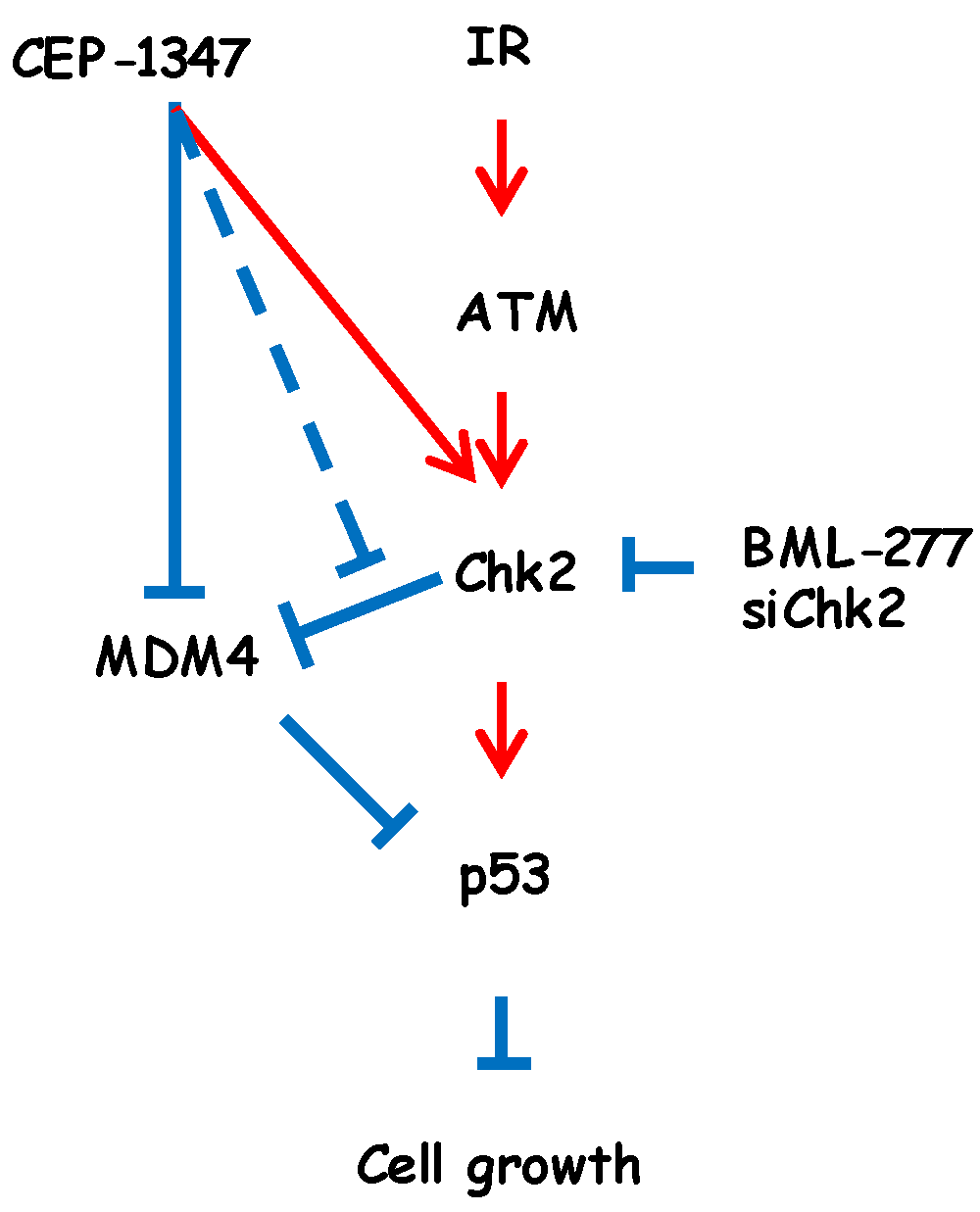
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Western Blot Analysis
4.4. Reverse Transcription–PCR Analysis
4.5. Gene Silencing by siRNA
4.6. Trypan Blue Dye Exclusion Assay
4.7. Colony Formation Assay
4.8. IR
4.9. Data Reproducibility and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marosi, C.; Hassler, M.; Roessler, K.; Reni, M.; Sant, M.; Mazza, E.; Vecht, C. Meningioma. Crit. Rev. Oncol. Hematol. 2008, 67, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Rohringer, M.; Sutherland, G.R.; Louw, D.F.; Sima, A.A. Incidence and clinicopathological features of meningioma. J. Neurosurg. 1989, 71, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro-Oncology 2022, 24, v1–v95. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Karschnia, P.; Young, J.S.; Dono, A.; Häni, L.; Sciortino, T.; Bruno, F.; Juenger, S.T.; Teske, N.; Morshed, R.A.; Haddad, A.F.; et al. Prognostic validation of a new classification system for extent of resection in glioblastoma: A report of the RANO resect group. Neuro-Oncol. 2023, 25, 940–954. [Google Scholar] [CrossRef]
- Goldbrunner, R.; Minniti, G.; Preusser, M.; Jenkinson, M.D.; Sallabanda, K.; Houdart, E.; von Deimling, A.; Stavrinou, P.; Lefranc, F.; Lund-Johansen, M.; et al. EANO guidelines for the diagnosis and treatment of meningiomas. Lancet Oncol. 2016, 17, e383–e391. [Google Scholar] [CrossRef]
- Khabibov, M.; Garifullin, A.; Boumber, Y.; Khaddour, K.; Fernandez, M.; Khamitov, F.; Khalikova, L.; Kuznetsova, N.; Kit, O.; Kharin, L. Signaling pathways and therapeutic approaches in glioblastoma multiforme (Review). Int. J. Oncol. 2022, 60, 69. [Google Scholar] [CrossRef]
- Goldbrunner, R.; Stavrinou, P.; Jenkinson, M.D.; Sahm, F.; Mawrin, C.; Weber, D.C.; Preusser, M.; Minniti, G.; Lund-Johansen, M.; Lefranc, F.; et al. EANO guideline on the diagnosis and management of meningiomas. Neuro-Oncol. 2021, 23, 1821–1834. [Google Scholar] [CrossRef]
- Patel, B.; Desai, R.; Pugazenthi, S.; Butt, O.H.; Huang, J.; Kim, A.H. Identification and Management of Aggressive Meningiomas. Front. Oncol. 2022, 12, 851758. [Google Scholar] [CrossRef]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Caspari, T. How to activate p53. Curr. Biol. 2000, 10, R315–R317. [Google Scholar] [CrossRef]
- Chen, L.; Gilkes, D.M.; Pan, Y.; Lane, W.S.; Chen, J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005, 24, 3411–3422. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.H.; Xu, Z.Y.; Mo, S.; Yuan, L.; Cheng, X.D.; Qin, J.J. Targeting MDMX for Cancer Therapy: Rationale, Strategies, and Challenges. Front. Oncol. 2020, 10, 1389. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef]
- Joachim, T.; Ram, Z.; Rappaport, Z.H.; Simon, M.; Schramm, J.; Wiestler, O.D.; von Deimling, A. Comparative analysis of the NF2, TP53, PTEN, KRAS, NRAS and HRAS genes in sporadic and radiation-induced human meningiomas. Int. J. Cancer 2001, 94, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Lechpammer, M. Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling. Cancers 2016, 8, 67. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef]
- Zhang, S.; Lou, J.; Li, Y.; Zhou, F.; Yan, Z.; Lyu, X.; Zhao, Y. Recent Progress and Clinical Development of Inhibitors that Block MDM4/p53 Protein-Protein Interactions. J. Med. Chem. 2021, 64, 10621–10640. [Google Scholar] [CrossRef]
- Miles, X.; Vandevoorde, C.; Hunter, A.; Bolcaen, J. MDM2/X Inhibitors as Radiosensitizers for Glioblastoma Targeted Therapy. Front. Oncol. 2021, 11, 703442. [Google Scholar] [CrossRef] [PubMed]
- Pellot Ortiz, K.I.; Rechberger, J.S.; Nonnenbroich, L.F.; Daniels, D.J.; Sarkaria, J.N. MDM2 Inhibition in the Treatment of Glioblastoma: From Concept to Clinical Investigation. Biomedicines 2023, 11, 1879. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2022, 79, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Mitobe, Y.; Nakagawa-Saito, Y.; Togashi, K.; Suzuki, S.; Sugai, A.; Matsuda, K.I.; Sonoda, Y.; Kitanaka, C.; Okada, M. CEP-1347 Targets MDM4 Protein Expression to Activate p53 and Inhibit the Growth of Glioma Cells. Anticancer. Res. 2022, 42, 4727–4733. [Google Scholar] [CrossRef]
- Mitobe, Y.; Suzuki, S.; Nakagawa-Saito, Y.; Togashi, K.; Sugai, A.; Sonoda, Y.; Kitanaka, C.; Okada, M. The Novel MDM4 Inhibitor CEP-1347 Activates the p53 Pathway and Blocks Malignant Meningioma Growth In Vitro and In Vivo. Biomedicines 2023, 11, 1967. [Google Scholar] [CrossRef] [PubMed]
- Maroney, A.C.; Glicksman, M.A.; Basma, A.N.; Walton, K.M.; Knight, E., Jr.; Murphy, C.A.; Bartlett, B.A.; Finn, J.P.; Angeles, T.; Matsuda, Y.; et al. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J. Neurosci. 1998, 18, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group PRECEPT Investigators. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 2007, 69, 1480–1490. [Google Scholar] [CrossRef]
- Goodfellow, V.S.; Loweth, C.J.; Ravula, S.B.; Wiemann, T.; Nguyen, T.; Xu, Y.; Todd, D.E.; Sheppard, D.; Pollack, S.; Polesskaya, O.; et al. Discovery, synthesis, and characterization of an orally bioavailable, brain penetrant inhibitor of mixed lineage kinase 3. J. Med. Chem. 2013, 56, 8032–8048. [Google Scholar] [CrossRef]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef]
- Togashi, K.; Okada, M.; Suzuki, S.; Sanomachi, T.; Seino, S.; Yamamoto, M.; Yamashita, H.; Kitanaka, C. Inhibition of Retinoblastoma Cell Growth by CEP1347 Through Activation of the P53 Pathway. Anticancer. Res. 2020, 40, 4961–4968. [Google Scholar] [CrossRef]
- Togashi, K.; Suzuki, S.; Mitobe, Y.; Nakagawa-Saito, Y.; Sugai, A.; Takenouchi, S.; Sugimoto, M.; Kitanaka, C.; Okada, M. CEP-1347 Dually Targets MDM4 and PKC to Activate p53 and Inhibit the Growth of Uveal Melanoma Cells. Cancers 2024, 16, 118. [Google Scholar] [CrossRef]
- Mitobe, Y.; Suzuki, S.; Nakagawa-Saito, Y.; Togashi, K.; Sugai, A.; Sonoda, Y.; Kitanaka, C.; Okada, M. Antagonizing MDM2 Overexpression Induced by MDM4 Inhibitor CEP-1347 Effectively Reactivates Wild-Type p53 in Malignant Brain Tumor Cells. Cancers 2023, 15, 4326. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394. [Google Scholar] [CrossRef]
- Melchionna, R.; Chen, X.B.; Blasina, A.; McGowan, C.H. Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat. Cell Biol. 2000, 2, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Abuetabh, Y.; Wu, H.H.; Chai, C.; Al Yousef, H.; Persad, S.; Sergi, C.M.; Leng, R. DNA damage response revisited: The p53 family and its regulators provide endless cancer therapy opportunities. Exp. Mol. Med. 2022, 54, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Fiscella, M.; Zhang, H.; Fan, S.; Sakaguchi, K.; Shen, S.; Mercer, W.E.; Vande Woude, G.F.; O’Connor, P.M.; Appella, E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 6048–6053. [Google Scholar] [CrossRef]
- Oliva-Trastoy, M.; Berthonaud, V.; Chevalier, A.; Ducrot, C.; Marsolier-Kergoat, M.C.; Mann, C.; Leteurtre, F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene 2007, 26, 1449–1458. [Google Scholar] [CrossRef]
- Lowe, J.; Cha, H.; Lee, M.O.; Mazur, S.J.; Appella, E.; Fornace, A.J., Jr. Regulation of the Wip1 phosphatase and its effects on the stress response. Front. Biosci. 2012, 17, 1480–1498. [Google Scholar] [CrossRef]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, L.; Guo, H.; Yang, J.; Jones, S.N.; Jochemsen, A.; Lu, X. Phosphorylation and degradation of MdmX is inhibited by Wip1 phosphatase in the DNA damage response. Cancer Res. 2009, 69, 7960–7968. [Google Scholar] [CrossRef]
- Hao, Q.; Chen, J.; Lu, H.; Zhou, X. The ARTS of p53-dependent mitochondrial apoptosis. J. Mol. Cell Biol. 2022, 14, mjac074. [Google Scholar] [CrossRef]
- Ranjan, A.; Iwakuma, T. Non-Canonical Cell Death Induced by p53. Int. J. Mol. Sci. 2016, 17, 2068. [Google Scholar] [CrossRef] [PubMed]
- Maroney, A.C.; Finn, J.P.; Connors, T.J.; Durkin, J.T.; Angeles, T.; Gessner, G.; Xu, Z.; Meyer, S.L.; Savage, M.J.; Greene, L.A.; et al. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J. Biol. Chem. 2001, 276, 25302–25308. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. Verteporfin inhibits oxidative phosphorylation and induces cell death specifically in glioma stem cells. FEBS J. 2020, 287, 2023–2036. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward | Reverse |
|---|---|---|
| MDM2 | GGTGCTGTAACCACCTCACA | TGAGTCCGATGATTCCTGCTG |
| CDKN1A | GGGATTTCTTCTGTTCAGGCG | TGGTAGAAATCTGTCATGCTGGT |
| ACTB | CCCATGCCATCCTGCGTCTG | CGTCATACTCCTGCTTGCTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitobe, Y.; Suzuki, S.; Nakamura, K.; Nakagawa-Saito, Y.; Takenouchi, S.; Togashi, K.; Sugai, A.; Sonoda, Y.; Kitanaka, C.; Okada, M. CEP-1347 Boosts Chk2-Mediated p53 Activation by Ionizing Radiation to Inhibit the Growth of Malignant Brain Tumor Cells. Int. J. Mol. Sci. 2024, 25, 9473. https://doi.org/10.3390/ijms25179473
Mitobe Y, Suzuki S, Nakamura K, Nakagawa-Saito Y, Takenouchi S, Togashi K, Sugai A, Sonoda Y, Kitanaka C, Okada M. CEP-1347 Boosts Chk2-Mediated p53 Activation by Ionizing Radiation to Inhibit the Growth of Malignant Brain Tumor Cells. International Journal of Molecular Sciences. 2024; 25(17):9473. https://doi.org/10.3390/ijms25179473
Chicago/Turabian StyleMitobe, Yuta, Shuhei Suzuki, Kazuki Nakamura, Yurika Nakagawa-Saito, Senri Takenouchi, Keita Togashi, Asuka Sugai, Yukihiko Sonoda, Chifumi Kitanaka, and Masashi Okada. 2024. "CEP-1347 Boosts Chk2-Mediated p53 Activation by Ionizing Radiation to Inhibit the Growth of Malignant Brain Tumor Cells" International Journal of Molecular Sciences 25, no. 17: 9473. https://doi.org/10.3390/ijms25179473