
An Inducible Luminescent System to Explore Parkinson’s Disease-Associated Genes

 , , and
, , and
Abstract
:1. Introduction
2. Results
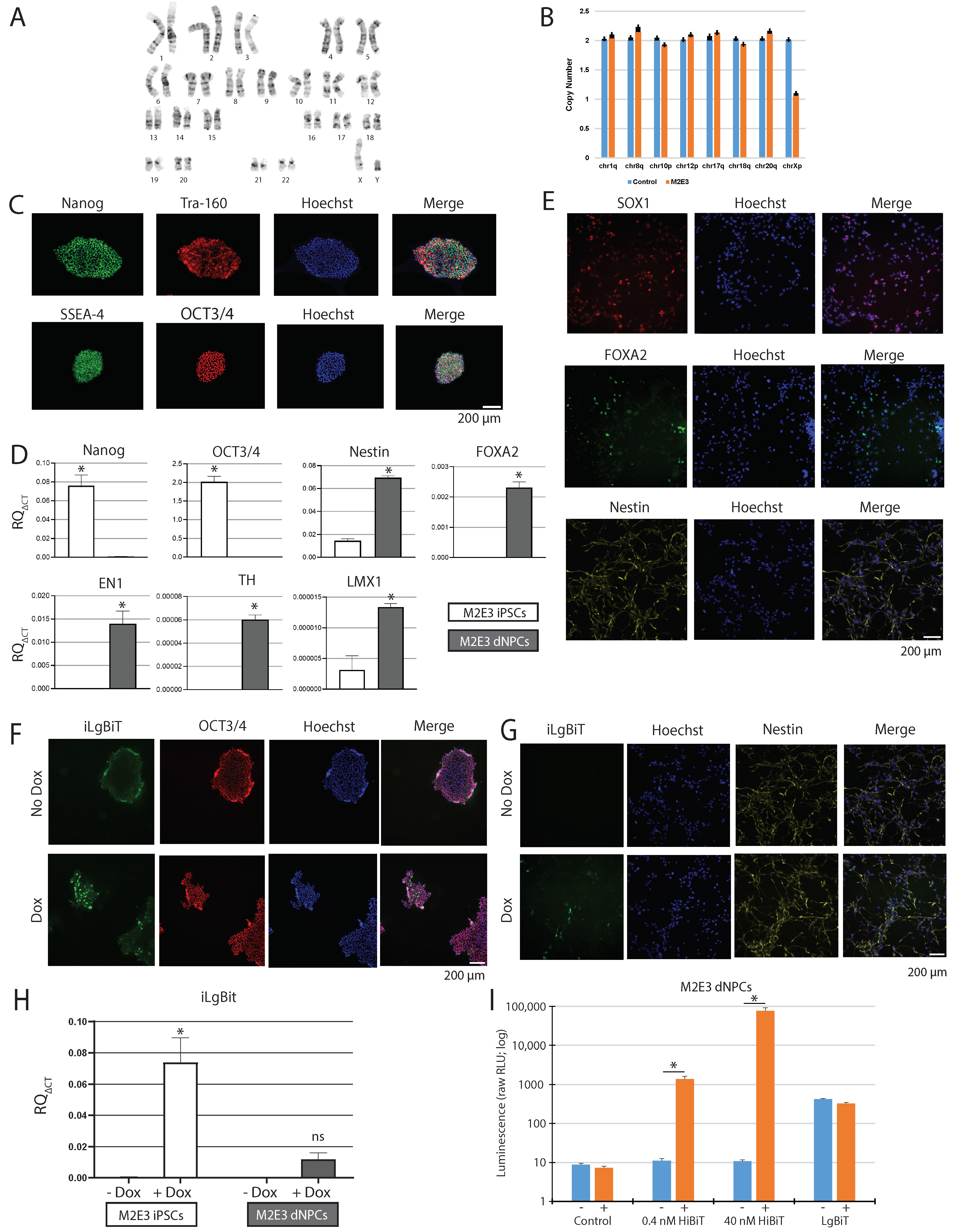
2.1. Engineering a Master Doxycycline-Inducible LgBiT iPSC Line
2.2. iLgBiT Copy Number Assessment and Clonal Validation
2.3. Confirmation of Heterozygosity
2.4. Expression of Active iLgBiT upon Doxycycline Treatment
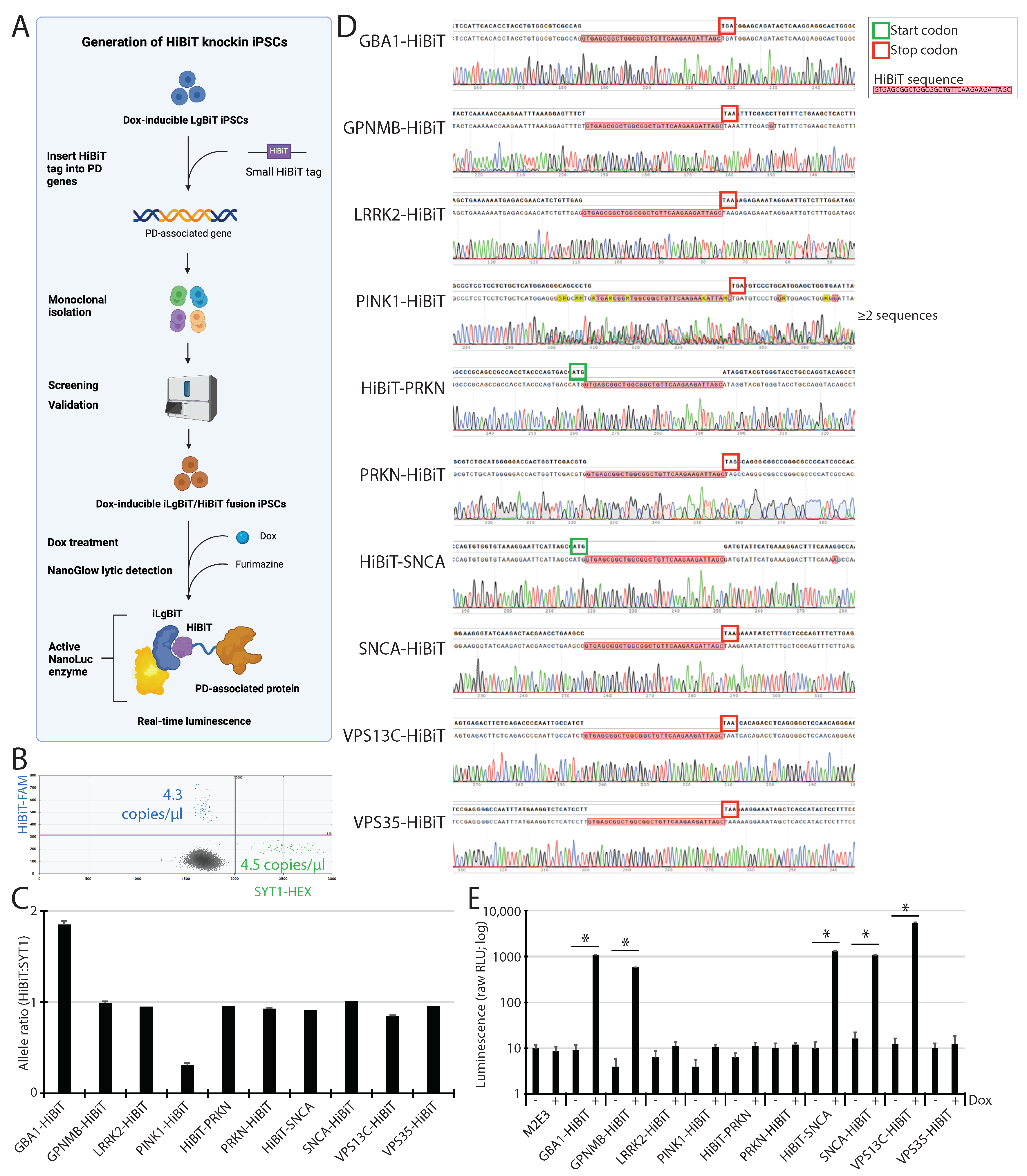
2.5. HiBiT Tagging to PD Genes into Master M2E3 iPSC Line
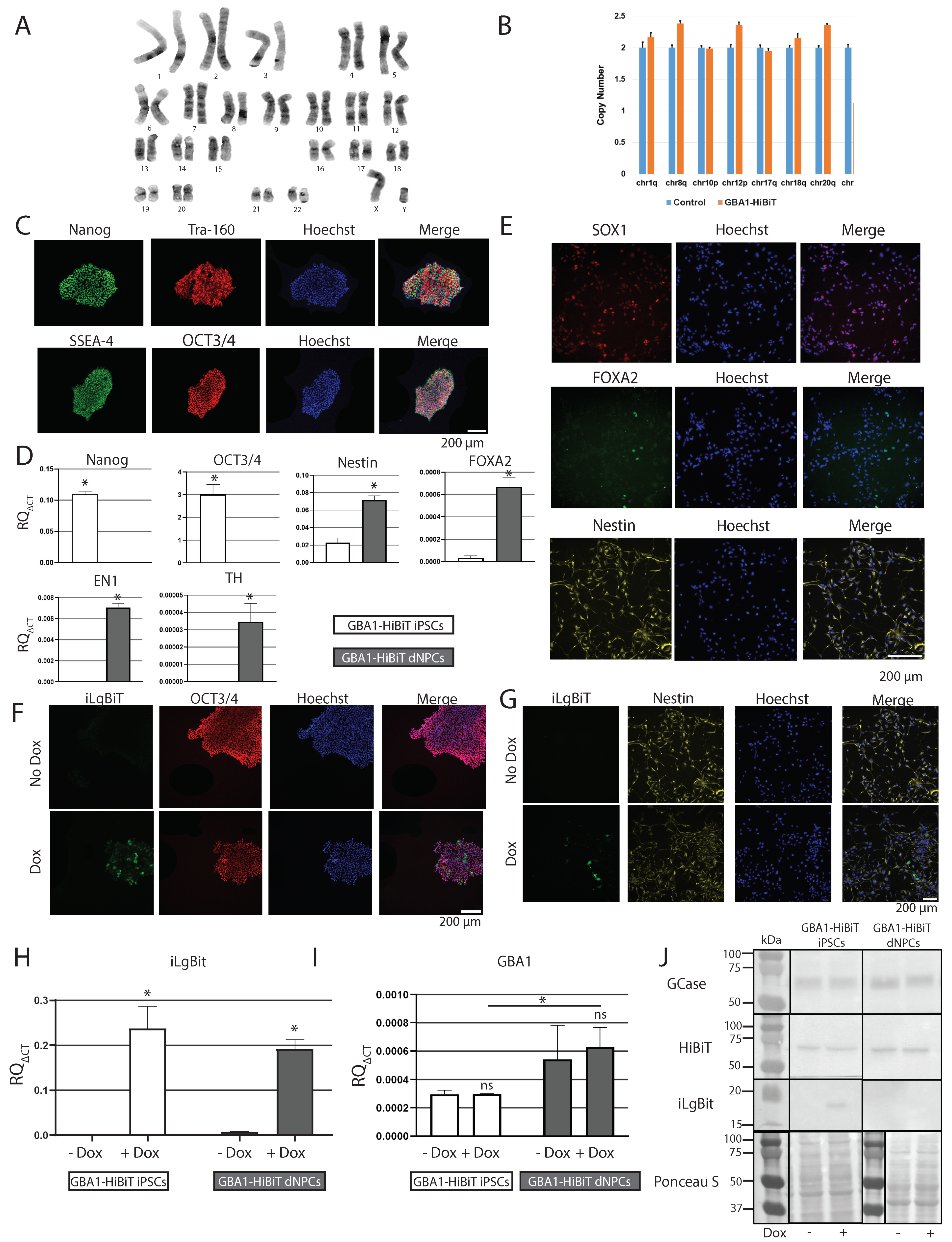
2.6. Characterization of GBA1-HiBiT iPSCs, dNPCs, and DNs
2.7. Validation of iLgBiT-HiBiT System with Pharmacological Chaperone Ambroxol
2.8. Off-Target Activity Analysis of CLYBL and GBA1 sgRNAs
3. Discussion
4. Materials and Methods
4.1. iPSC Culture
4.2. Karyotyping
4.3. Generating a Doxycycline-Inducible LgBiT iPSC Line
4.4. Generation of HiBiT iPSC Lines
4.5. Doxycycline-Induction of iLgBiT Expression
4.6. RT-qPCR
4.7. Induction and Differentiation of iPSC-Derived Dopaminergic Progenitors and Neurons
4.8. Ambroxol Treatment
4.9. Western Blot
4.10. Luminescence Detection
4.11. GCase Enzyme Activity Assay
4.12. Immunocytochemistry Analysis
4.13. Off-Target Analysis of LgBiT and HiBiT Insertions
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lebouvier, T.; Chaumette, T.; Paillusson, S.; Duyckaerts, C.; Bruley des Varannes, S.; Neunlist, M.; Derkinderen, P. The second brain and Parkinson’s disease. Eur. J. Neurosci. 2009, 30, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef]
- Al-Azzawi, Z.A.M.; Arfaie, S.; Gan-Or, Z. GBA1 and The Immune System: A Potential Role in Parkinson’s Disease? J. Parkinsons Dis. 2022, 12, S53–S64. [Google Scholar] [CrossRef]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Schreij, A.M.; Chaineau, M.; Ruan, W.; Lin, S.; Barker, P.A.; Fon, E.A.; McPherson, P.S. LRRK2 localizes to endosomes and interacts with clathrin-light chains to limit Rac1 activation. EMBO Rep. 2015, 16, 79–86. [Google Scholar] [CrossRef]
- Ayoubi, R.; Ryan, J.; Biddle, M.S.; Alshafie, W.; Fotouhi, M.; Bolivar, S.G.; Ruiz Moleon, V.; Eckmann, P.; Worrall, D.; McDowell, I.; et al. Scaling of an antibody validation procedure enables quantification of antibody performance in major research applications. Elife 2023, 12, e91645. [Google Scholar] [CrossRef]
- Laflamme, C.; McKeever, P.M.; Kumar, R.; Schwartz, J.; Kolahdouzan, M.; Chen, C.X.; You, Z.; Benaliouad, F.; Gileadi, O.; McBride, H.M.; et al. Implementation of an antibody characterization procedure and application to the major ALS/FTD disease gene C9ORF72. Elife 2019, 8, e48363. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Pluckthun, A. Reproducibility: Standardize antibodies used in research. Nature 2015, 518, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; McDonald, D.; Blain, A.; Mossman, E.; Atkin, K.; Marusich, M.F.; Capaldi, R.; Bone, L.; Smith, A.; Filby, A.; et al. Parkinson’s disease neurons exhibit alterations in mitochondrial quality control proteins. NPJ Parkinsons Dis. 2023, 9, 120. [Google Scholar] [CrossRef]
- Lackner, D.H.; Carre, A.; Guzzardo, P.M.; Banning, C.; Mangena, R.; Henley, T.; Oberndorfer, S.; Gapp, B.V.; Nijman, S.M.B.; Brummelkamp, T.R.; et al. A generic strategy for CRISPR-Cas9-mediated gene tagging. Nat. Commun. 2015, 6, 10237. [Google Scholar] [CrossRef] [PubMed]
- Moriya, H. Quantitative nature of overexpression experiments. Mol. Biol. Cell 2015, 26, 3932–3939. [Google Scholar] [CrossRef]
- Jarvik, J.W.; Telmer, C.A. Epitope tagging. Annu. Rev. Genet. 1998, 32, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Dewari, P.S.; Southgate, B.; McCarten, K.; Monogarov, G.; O’Duibhir, E.; Quinn, N.; Tyrer, A.; Leitner, M.C.; Plumb, C.; Kalantzaki, M.; et al. An efficient and scalable pipeline for epitope tagging in mammalian stem cells using Cas9 ribonucleoprotein. Elife 2018, 7, e35069. [Google Scholar] [CrossRef]
- Fan, F.; Wood, K.V. Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 2007, 5, 127–136. [Google Scholar] [CrossRef]
- Schwinn, M.K.; Machleidt, T.; Zimmerman, K.; Eggers, C.T.; Dixon, A.S.; Hurst, R.; Hall, M.P.; Encell, L.P.; Binkowski, B.F.; Wood, K.V. CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem. Biol. 2018, 13, 467–474. [Google Scholar] [CrossRef]
- Schwinn, M.K.; Steffen, L.S.; Zimmerman, K.; Wood, K.V.; Machleidt, T. A Simple and Scalable Strategy for Analysis of Endogenous Protein Dynamics. Sci. Rep. 2020, 10, 8953. [Google Scholar] [CrossRef]
- Chen, C.X.; Abdian, N.; Maussion, G.; Thomas, R.A.; Demirova, I.; Cai, E.; Tabatabaei, M.; Beitel, L.K.; Karamchandani, J.; Fon, E.A.; et al. A Multistep Workflow to Evaluate Newly Generated iPSCs and Their Ability to Generate Different Cell Types. Methods Protoc. 2021, 4, 50. [Google Scholar] [CrossRef]
- Cerbini, T.; Funahashi, R.; Luo, Y.; Liu, C.; Park, K.; Rao, M.; Malik, N.; Zou, J. Transcription activator-like effector nuclease (TALEN)-mediated CLYBL targeting enables enhanced transgene expression and one-step generation of dual reporter human induced pluripotent stem cell (iPSC) and neural stem cell (NSC) lines. PLoS ONE 2015, 10, e0116032. [Google Scholar] [CrossRef]
- Alfradique-Dunham, I.; Al-Ouran, R.; von Coelln, R.; Blauwendraat, C.; Hill, E.; Luo, L.; Stillwell, A.; Young, E.; Kaw, A.; Tan, M.; et al. Genome-Wide Association Study Meta-Analysis for Parkinson Disease Motor Subtypes. Neurol. Genet. 2021, 7, e557. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N. Alpha-synuclein structure, functions, and interactions. J. Res. Med. Sci. 2016, 21, 29. [Google Scholar] [CrossRef]
- Chaugule, V.K.; Burchell, L.; Barber, K.R.; Sidhu, A.; Leslie, S.J.; Shaw, G.S.; Walden, H. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 2011, 30, 2853–2867. [Google Scholar] [CrossRef]
- Burchell, L.; Chaugule, V.K.; Walden, H. Small, N-terminal tags activate Parkin E3 ubiquitin ligase activity by disrupting its autoinhibited conformation. PLoS ONE 2012, 7, e34748. [Google Scholar] [CrossRef] [PubMed]
- Maric, G.; Rose, A.A.; Annis, M.G.; Siegel, P.M. Glycoprotein non-metastatic b (GPNMB): A metastatic mediator and emerging therapeutic target in cancer. Onco Targets Ther. 2013, 6, 839–852. [Google Scholar] [CrossRef]
- Cresto, N.; Gardier, C.; Gaillard, M.C.; Gubinelli, F.; Roost, P.; Molina, D.; Josephine, C.; Dufour, N.; Auregan, G.; Guillermier, M.; et al. The C-Terminal Domain of LRRK2 with the G2019S Substitution Increases Mutant A53T alpha-Synuclein Toxicity in Dopaminergic Neurons In Vivo. Int. J. Mol. Sci. 2021, 22, 6760. [Google Scholar] [CrossRef]
- Kakade, P.; Ojha, H.; Raimi, O.G.; Shaw, A.; Waddell, A.D.; Ault, J.R.; Burel, S.; Brockmann, K.; Kumar, A.; Ahangar, M.S.; et al. Mapping of a N-terminal alpha-helix domain required for human PINK1 stabilization, Serine228 autophosphorylation and activation in cells. Open Biol. 2022, 12, 210264. [Google Scholar] [CrossRef]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.Y.; Whitworth, A.J.; Schapira, A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef]
- Williams, D.; Glasstetter, L.M.; Jong, T.T.; Kapoor, A.; Zhu, S.; Zhu, Y.; Gehrlein, A.; Vocadlo, D.J.; Jagasia, R.; Marugan, J.J.; et al. Development of quantitative high-throughput screening assays to identify, validate, and optimize small-molecule stabilizers of misfolded beta-glucocerebrosidase with therapeutic potential for Gaucher disease and Parkinson’s disease. bioRxiv 2024. [Google Scholar] [CrossRef]
- Hamdan, A.; Ewing, A. Unravelling the tumour genome: The evolutionary and clinical impacts of structural variants in tumourigenesis. J. Pathol. 2022, 257, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P.; Lee, G.; Malani, N.; Setty, M.; Riviere, I.; Tirunagari, L.M.; Kadota, K.; Roth, S.L.; Giardina, P.; Viale, A.; et al. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 73–78. [Google Scholar] [CrossRef]
- Deneault, E.; Chaineau, M.; Nicouleau, M.; Castellanos Montiel, M.J.; Franco Flores, A.K.; Haghi, G.; Chen, C.X.; Abdian, N.; Shlaifer, I.; Beitel, L.K.; et al. A streamlined CRISPR workflow to introduce mutations and generate isogenic iPSCs for modeling amyotrophic lateral sclerosis. Methods 2022, 203, 297–310. [Google Scholar] [CrossRef]
- Deneault, E.; Faheem, M.; White, S.H.; Rodrigues, D.C.; Sun, S.; Wei, W.; Piekna, A.; Thompson, T.; Howe, J.L.; Chalil, L.; et al. CNTN5−/+ or EHMT2−/+ human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. Elife 2019, 8, e40092. [Google Scholar] [CrossRef]
- Deneault, E.; White, S.H.; Rodrigues, D.C.; Ross, P.J.; Faheem, M.; Zaslavsky, K.; Wang, Z.; Alexandrova, R.; Pellecchia, G.; Wei, W.; et al. Complete Disruption of Autism-Susceptibility Genes by Gene Editing Predominantly Reduces Functional Connectivity of Isogenic Human Neurons. Stem Cell Rep. 2018, 11, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Chan, A.H.; Judge, L.M.; Yoo, J.; Huang, M.; Nguyen, T.D.; Lizarraga, P.P.; So, P.L.; Conklin, B.R. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods 2014, 11, 291–293. [Google Scholar] [CrossRef]
- Mohamed, N.V.; Sirois, J.; Ramamurthy, J.; Mathur, M.; Lepine, P.; Deneault, E.; Maussion, G.; Nicouleau, M.; Chen, C.X.; Abdian, N.; et al. Midbrain organoids with an SNCA gene triplication model key features of synucleinopathy. Brain Commun. 2021, 3, fcab223. [Google Scholar] [CrossRef]
- Ross, P.J.; Zhang, W.B.; Mok, R.S.F.; Zaslavsky, K.; Deneault, E.; D’Abate, L.; Rodrigues, D.C.; Yuen, R.K.C.; Faheem, M.; Mufteev, M.; et al. Synaptic Dysfunction in Human Neurons With Autism-Associated Deletions in PTCHD1-AS. Biol. Psychiatry 2020, 87, 139–149. [Google Scholar] [CrossRef]
- Zaslavsky, K.; Zhang, W.B.; McCready, F.P.; Rodrigues, D.C.; Deneault, E.; Loo, C.; Zhao, M.; Ross, P.J.; El Hajjar, J.; Romm, A.; et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 2019, 22, 556–564. [Google Scholar] [CrossRef]
- Saeed, A.F.; Wang, R.; Ling, S.; Wang, S. Antibody Engineering for Pursuing a Healthier Future. Front. Microbiol. 2017, 8, 495. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Tabak, L.A. Policy: NIH plans to enhance reproducibility. Nature 2014, 505, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Higashi, S.; Moore, D.J.; Colebrooke, R.E.; Biskup, S.; Dawson, V.L.; Arai, H.; Dawson, T.M.; Emson, P.C. Expression and localization of Parkinson’s disease-associated leucine-rich repeat kinase 2 in the mouse brain. J. Neurochem. 2007, 100, 368–381. [Google Scholar] [CrossRef]
- Stichel, C.C.; Augustin, M.; Kuhn, K.; Zhu, X.R.; Engels, P.; Ullmer, C.; Lubbert, H. Parkin expression in the adult mouse brain. Eur. J. Neurosci. 2000, 12, 4181–4194. [Google Scholar] [CrossRef]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef]
- Kamiya, K.; Kaneda, M.; Sugawara, T.; Mazaki, E.; Okamura, N.; Montal, M.; Makita, N.; Tanaka, M.; Fukushima, K.; Fujiwara, T.; et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J. Neurosci. 2004, 24, 2690–2698. [Google Scholar] [CrossRef]
- Luo, H.; Zheng, R.; Zhao, Y.; Wu, J.; Li, J.; Jiang, F.; Chen, D.N.; Zhou, X.T.; Li, J.D. A dominant negative FGFR1 mutation identified in a Kallmann syndrome patient. Gene 2017, 621, 1–4. [Google Scholar] [CrossRef]
- Shojima, Y.; Ogata, J.; Tsunemi, T.; Imai, Y.; Hattori, N. Measurement of GCase Activity in Cultured Cells. Methods Mol. Biol. 2021, 2322, 47–52. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone | LgBiT-FAM (copies/µL) | SYT1-HEX (copies/µL) | Ratio iLgBiT:SYT1 | Clone | LgBiT-FAM (copies/µL) | SYT1-HEX (copies/µL) | Ratio iLgBiT:SYT1 | Clone | LgBiT-FAM (copies/µL) | SYT1-HEX (copies/µL) | Ratio iLgBiT:SYT1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M1C6.1 | 525 | 193 | 2.7 | M2E3.v3-1 | 99 | 192 | 0.5 | M1G4.v3-1 | 78 | 175 | 0.4 | |
| M1C6.2 | 326 | 88 | 3.7 | M2E3.v3-2 | 29 | 60 | 0.5 | M1G4.v3-2 | 936 | 377 | 2.5 | |
| M1C6.3 | 295 | 89 | 3.3 | M2E3.v3-3 | 135 | 271 | 0.5 | M1G4.v3-3 | 104 | 213 | 0.5 | |
| M1C6.4 | 615 | 231 | 2.7 | M2E3.v3-4 | 96 | 146 | 0.7 | M1G4.v3-4 | 67 | 158 | 0.4 | |
| M1C6.5 | 612 | 191 | 3.2 | M2E3.v3-5 | 75 | 155 | 0.5 | M1G4.v3-5 | 197 | 466 | 0.4 | |
| M1C6.6 | 425 | 120 | 3.5 | M2E3.v3-6 | 38 | 72 | 0.5 | M1G4.v3-6 | 181 | 413 | 0.4 | |
| M1C6.7 | 671 | 207 | 3.2 | M2E3.v3-7 | 64 | 122 | 0.5 | M1G4.v3-7 | 130 | 307 | 0.4 | |
| M1C6.8 | 409 | 119 | 3.4 | M2E3.v3-8 | 56 | 146 | 0.4 | M1G4.v3-8 | 25 | 52 | 0.5 | |
| M1C6.9 | 499 | 146 | 3.4 | M2E3.v3-9 | 78 | 152 | 0.5 | M1G4.v3-9 | 130 | 285 | 0.5 | |
| M1C6.11 | 523 | 169 | 3.1 | M2E3.v3-10 | 96 | 204 | 0.5 | M1G4.v3-10 | 111 | 231 | 0.5 | |
| M1C6.12 | 464 | 129 | 3.6 | M2E3.v3-11 | 95 | 209 | 0.5 | M1G4.v3-11 | 112 | 253 | 0.4 | |
| M1C6.13 | 525 | 187 | 2.8 | M2E3.v3-12 | 88 | 177 | 0.5 | M1G4.v3-12 | 243 | 506 | 0.5 | |
| M1C6.14 | 305 | 105 | 2.9 | M2E3.v3-13 | 73 | 131 | 0.6 | M1G4.v3-13 | 201 | 458 | 0.4 | |
| M1C6.15 | 216 | 246 | 0.9 | M2E3.v3-14 | 22 | 56 | 0.4 | M1G4.v3-14 | 1663 | 633 | 2.6 | |
| M2E3.v3-15 | 109 | 241 | 0.5 | M1G4.v3-15 | 89 | 32 | 2.8 | |||||
| M2E3.v3-16 | 127 | 287 | 0.4 | M1G4.v3-16 | 199 | 482 | 0.4 | |||||
| Ave | 3.0 | 0.5 | 0.9 | |||||||||
| SD | 0.7 | 0.07 | 0.9 |
| Oligonucleotide Name | Oligonucleotide Sequence |
|---|---|
| CLYBL-sgRNA | ATGTTGGAAGGATGAGGAAA |
| CLYBL-LgBiT-taqF | CATCTGATCGGCAGAGAGC |
| CLYBL-LgBiT-taqR | ACAAGGTGGCGTCAGTTC |
| SYT1-Ftaq | AGCCATAGTCGCAGTCCT |
| SYT1-Rtaq | ACCTGATCTTTCATCGTCTTCC |
| CLYBL-LgBiT-FAM | CCA+GA+C+C+GGA |
| SYT1-wtHEX | AA+GAA+GAA+G+G+GA |
| CLY5-F2 | TCCTACTGGAGACACAGGTCC |
| Neo-R2 | TCCACGTCACCGCATGTTAG |
| CLY5-F1 | GCTTGCACGTCTGGAACTCT |
| Cre-R1 | GGCAAACAACAGATGGCTGG |
| CLYwt-F2 | AAAACAGCATGACTGGTGGC |
| CLYwt-R2 | ACCTGACGTTTTTCTACTGGGA |
| M13rev | CAGGAAACAGCTATGAC |
| LNCX | AGCTCGTTTAGTGAACCGTCAGATC |
| Gene Fusion | Transcript ID | Oligonucleotide Name | Oligonucleotide Sequence |
|---|---|---|---|
| HiBiT-SNCA | ENST00000394991.8 | gRNA | GTAAAGGAATTCATTAGCCA |
| ssODN | TTCTCATTCAAAGTGTATTTTATGTTTTCCAGTGTGGTGTAAAGGAATTCATTAGCCATGGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCGATGTATTCATGAAAGGACTTTCAAAGGCCAAGGAGGGAGTTGTGGCTGCTGCTGAGAAA | ||
| Ftaq | TGTATTTTATGTTTTCCAGTGTGGT | ||
| Rtaq | GCCACACCCTGTTTGGT | ||
| wt-HEX | CA+T+G+G+AT+G+TATTCA | ||
| HiBiT-FAM | CA+T+GG+TGA+G+CG | ||
| Fseq | TCCGTGGTTAGGTGGCTAGA | ||
| Rseq | CCATCACTCATGAACAAGCACC | ||
| SNCA-HiBiT | ENST00000394991.8 | gRNA | TGGGAGCAAAGATATTTCTT |
| ssODN | GTGCTGTCTTTTTGATTTTTCTAATATTAGGAAGGGTATCAAGACTACGAACCTGAAGCCGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTAAGAAATATCTTTGCTCCCAGTTTCTTGAGATCTGCTGACAGATGTTCCATCCTGTACAA | ||
| Ftaq | TTAGGAAGGGTATCAAGACTACG | ||
| Rtaq | GGCACATTGGAACTGAGC | ||
| wt-HEX | CT+G+AAG+C+C+T+AAG | ||
| HiBiT-FAM | CTG+G+CG+G+CTG | ||
| Fseq | GTGCATCCGGATCAGAACCTA | ||
| Rseq | CAGTGAAAGGGAAGCACCGA | ||
| LRRK2-HiBiT | ENST00000298910.12 | gRNA | CTGTTGAGTAAGAGAGAAAT |
| ssODN | GAAAAACACATTGAAGTGAGAAAAGAATTAGCTGAAAAAATGAGACGAACATCTGTTGAGGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTAAGAGAGAAATACGAATTGTCTTTGGATAGGAAAATTATTCTCTCCTCTTGTAAATATTTATTTTAAA | ||
| Ftaq | ATCAATCTTCCACATGAAGTGC | ||
| Rtaq | ATGTGAGTACCCTTTCCATGT | ||
| wt-HEX | T+C+T+GTT+G+A+GTAAGA | ||
| HiBiT-FAM | CGG+CTGG+CGG | ||
| Fseq | ACTAAAAATACATGAGCCAAACTGA | ||
| Rseq | ACCTCCATTACAGACAAGAAAACA | ||
| PINK1-HiBiT | ENST00000321556.5 | gRNA | GCTCCATGCAGGGACATCAC |
| ssODN | TCTGCCAGGCAGCCCTCCTCCTCTGCTCATGGAGGGCAGCCCTGGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTGATGTCCCTGCATGGAGCTGGTGAATTACTAAAAGAACATGGCATCCTCTGTG | ||
| Ftaq | CTGGAGTGTGAAACGCTCT | ||
| Rtaq | CACAGACCATCACGACACA | ||
| wt-HEX | AG+CC+CTGT+G+ATG | ||
| HiBiT-FAM | A+G+ATTA+G+C+T+GATGT | ||
| Fseq | AGACCCTCACTAACAAAGCAGG | ||
| Rseq | TTCTTCCATTTGCCAAGCCC | ||
| GPNMB-HiBiT | ENST00000258733.9 | gRNA | AAAAGTGAGCTTCAGAAACA |
| ssODN | TTCCCGGGAAACCAGGAAAAGGATCCGCTACTCAAAAACCAAGAATTTAAAGGAGTTTCTGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTAAATTTCGACGTTGTTTCTGAAGCTCACTTTTCAGTGCCATTGATGTGAGATGTGCTGGAGTGGCTATTAACCTT | ||
| Ftaq | CCGGGAAACCAGGAAAAGG | ||
| Rtaq | TCAACTTCCCCAAACCACAA | ||
| wt-HEX | A+G+G+A+GTT+T+CTTAAA | ||
| HiBiT-FAM | CG+GC+TGG+CGG | ||
| Fseq | CCAGTGTCTTGCAAACTGTCAA | ||
| Rseq | GCTGCCTGCAGTATAATCCCT | ||
| VPS35-HiBiT | ENST00000299138.12 | gRNA | GAAGGTCTCATCCTTTAAAA |
| ssODN | TTGCGCTTGCGGCGGGAATCACCAGAATCCGAGGGGCCAATTTATGAAGGTCTCATCCTTGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTAAAAAGGAAATAGCTCACCATACTCCTTTCCATGTACATCCAGTGAGGGTTTTATTACGCT | ||
| Ftaq | CGGGAATCACCAGAATCCGA | ||
| Rtaq | AGGCACAATCTATGGAAGGG | ||
| wt-HEX | T+C+T+CAT+C+C+TTTAAA | ||
| HiBiT-FAM | CAA+G+A+A+GATTA+G+CT | ||
| Fseq | GAGTGTAACAGAAGCTCCTCA | ||
| Rseq | CAGTCATGCTACTTGGGGTGA | ||
| GBA-HiBiT | ENST00000327247.9 | gRNA | ACCTGTGGCGTCGCCAGTGA |
| ssODN | GGCTTCCTGGAGACAATCTCACCTGGCTACTCCATTCACACCTACCTGTGGCGTCGCCAGGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTGATGGAGCAGATACTCAAGGAGGCACTGGGCTCAGCCTGGGCATTAAAGGGACAGAGTCAGC | ||
| Ftaq | ATTCACACCTACCTGTGGC | ||
| Rtaq | CCTGCTGTGCCCTCTTTAG | ||
| wt-HEX | CG+C+CAG+T+GATG | ||
| HiBiT-FAM | AT+TA+G+C+T+GAT+GGAG | ||
| Fseq | CTAAACCGGTGAGGGCAATG | ||
| Rseq | GGGGAAAGTGAGTCACCCAAA | ||
| VPS13C-HiBiT | ENST00000644861.2 | gRNA | CCTGAGGTCTGTGATTAAGA |
| ssODN | CAGCAGCAAAAATTGATGAAGCAGTCATCAGTGAGACTTCTCAGACCCCAATTGCCATCTGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTAATCACAGACCTCAGGGGCTCCAACAGGGAGAAAAAACAATCACTGGTCTTGTCTAT | ||
| Ftaq | AGTGAGACTTCTCAGACCCC | ||
| Rtaq | AGCAAGATAAAGCAGAGTGACTT | ||
| wt-HEX | TG+C+CA+T+C+T+TAATC | ||
| HiBiT-FAM | CGG+CTGG+CGG | ||
| Fseq | TATGCTGTGGACTCAGTCGG | ||
| Rseq | TCTGACCCATTTGGGTGGTG | ||
| HiBiT-PRKN | ENST00000366898.6 | gRNA | ACCTACCCAGTGACCATGAT |
| ssODN | GCGCATGGGCCTGTTCCTGGCCCGCAGCCGCCACCTACCCAGTGACCATGGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCATAGGTACGTGGGTACCTGCCAGGTACAGCCTCTCTGCGCCGCCCCACGCC | ||
| Ftaq | CATGGGCCTGTTCCTGG | ||
| Rtaq | GTCATTGACAGTTGGCACC | ||
| wt-HEX | A+C+CAT+G+A+TA+GGT | ||
| HiBiT-FAM | CA+TG+GT+G+AG+CG | ||
| Fseq | CGGTGACGTAAGATTGCTGG | ||
| Rseq | GGCTTCGAACGCACACACT | ||
| PRKN-HiBiT | ENST00000366898.6 | gRNA | ACTGGTTCGACGTGTAGCCA |
| ssODN | GAACTGTGGCTGCGAGTGGAACCGCGTCTGCATGGGGGACCACTGGTTCGACGTGGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTAGCCAGGGCGGCCGGGCGCCCCATCGCCACATCCTGGGGGAGCAT | ||
| Ftaq | GAACTGTGGCTGCGAGT | ||
| Rtaq | AGAAAATGAAGGTAGACACTGGG | ||
| wt-HEX | TC+G+A+C+GT+GTA+GC | ||
| HiBiT-FAM | CG+GC+TGG+CGG | ||
| Fseq | TCCCGACAAAAGTGACATGCT | ||
| Rseq | TTTGTGTCATCCGGAGGCTG |
| Gene | Sequence | Reference | Vendor |
|---|---|---|---|
| Nanog | n/a | Hs02387400_g1 | Applied Biosystems |
| OCT3/4 | n/a | Hs04260367_gH | Applied Biosystems |
| Nestin | n/a | Hs04187831_g1 | Applied Biosystems |
| FOXA2 | n/a | Hs00232764_m1 | Applied Biosystems |
| Nurr1 (NR4A2) | n/a | Hs01117527_g1 | Applied Biosystems |
| GBA | n/a | Hs.PT.58.40746061 | Applied Biosystems |
| GAPDH | n/a | Hs02786624_g1 | Applied Biosystems |
| ACTB | n/a | Hs01060665_g1 | Applied Biosystems |
| GBA | n/a | Hs.PT.58.40746061 | IDT |
| ACTB | n/a | Hs.PT.39a.22214847 | IDT |
| GAPDH | n/a | Hs.PT.39a.22214836 | IDT |
| GBA-HiBiT-F | GACAATCTCACCTGGCTACTC | n/a | IDT |
| GBA-HiBiT-R | CTCCTTGAGTATCTGCTCCATC | n/a | IDT |
| GBA-HiBiT probe | TGGCGGCTGTTCAAGAAGATTAGCT | n/a | IDT |
| LgBiT-F | CCGATCAGATGGCGCAAATA | n/a | IDT |
| LgBiT-R | GGTTCCGTAGGGCAGAATTAC | n/a | IDT |
| Cell type | Antibody | Catalog | Dilution |
|---|---|---|---|
| iPSC | Nanog | Abcam Ab21624 | 1:200 |
| TRA1-60 | STEMCELL Technologies #60064 | 1:200 IF | |
| OCT3/4 | Santa Cruz SC-8628 | 1:2000 IF | |
| SSEA-4 | Santa Cruz SC-21704 | 1:500 IF | |
| dNPC | Nestin | Abcam ab92391 | 1:500 IF |
| SOX1 | R&D AF3369 | 1:500 IF | |
| FOXA2 | R&D AF2400 | 1:500 IF | |
| LgBiT | Promega N7100 | 1:100 IF 1:1000 WB | |
| HiBiT | Promega N7200 | 1:100 IF 1:1000 WB |
| CLYBL-17-F | ACTTACCAGCAACCTCGGTG |
| CLYBL-17-R | CATAGCCCCAGTCTGTGCAA |
| CLYBL-6-F | TGTGTCAGGGCTAAGAGTGC |
| CLYBL-6-R | TGACAGGCACATGGGGTTAG |
| CLYBL-10-F | GGAATCGTTTGCAGCCAGTG |
| CLYBL-10-R | CTACCCATCGCTCGGTCAC |
| GBA-20-F | TGACCTCTGGCTTCCATCAAG |
| GBA-20-R | GACCTATCTCTCAGAGCTGCC |
| GBA-1-F | GAAGCAGATGACACCTTGGC |
| GBA-1-R | TAGTTGGATGAAGGCTCTGGC |
| GBA-5-F | GAGCTGACGCTATTCGGTTTG |
| GBA-5-R | TTGGATGTCTGCCCCTTCG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandy, A.; Maussion, G.; Al-Habyan, S.; Nicouleau, M.; You, Z.; Chen, C.X.-Q.; Abdian, N.; Aprahamian, N.; Krahn, A.I.; Larocque, L.; et al. An Inducible Luminescent System to Explore Parkinson’s Disease-Associated Genes. Int. J. Mol. Sci. 2024, 25, 9493. https://doi.org/10.3390/ijms25179493
Gandy A, Maussion G, Al-Habyan S, Nicouleau M, You Z, Chen CX-Q, Abdian N, Aprahamian N, Krahn AI, Larocque L, et al. An Inducible Luminescent System to Explore Parkinson’s Disease-Associated Genes. International Journal of Molecular Sciences. 2024; 25(17):9493. https://doi.org/10.3390/ijms25179493
Chicago/Turabian StyleGandy, Anelya, Gilles Maussion, Sara Al-Habyan, Michael Nicouleau, Zhipeng You, Carol X.-Q. Chen, Narges Abdian, Nathalia Aprahamian, Andrea I. Krahn, Louise Larocque, and et al. 2024. "An Inducible Luminescent System to Explore Parkinson’s Disease-Associated Genes" International Journal of Molecular Sciences 25, no. 17: 9493. https://doi.org/10.3390/ijms25179493