
Prediction of Multiple Degenerative Diseases Based on DNA Methylation in a Co-Physiology Mechanisms Perspective

Abstract
:1. Introduction
2. Results
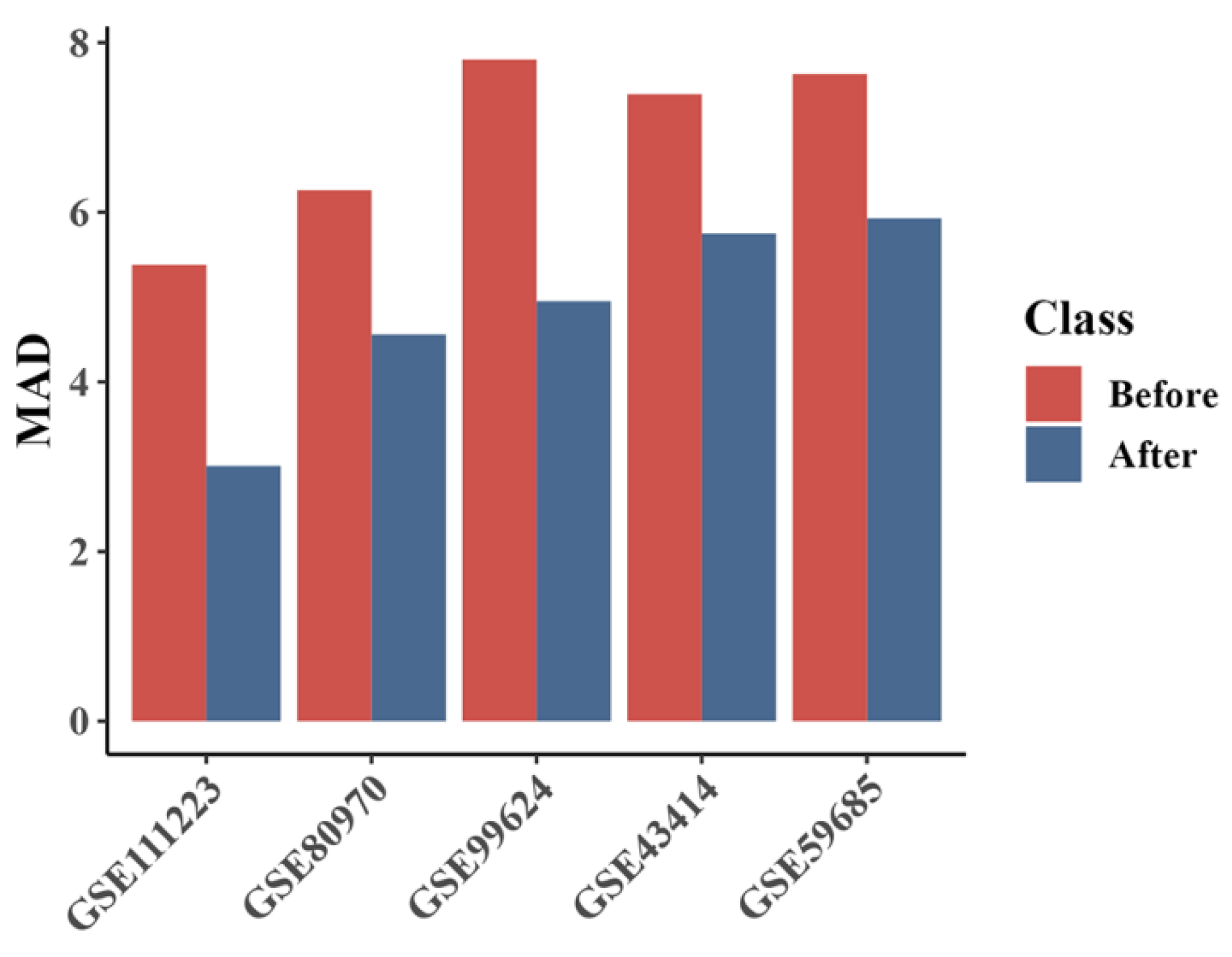
2.1. Validation and Performance Enhancement with Selected CpG Sites
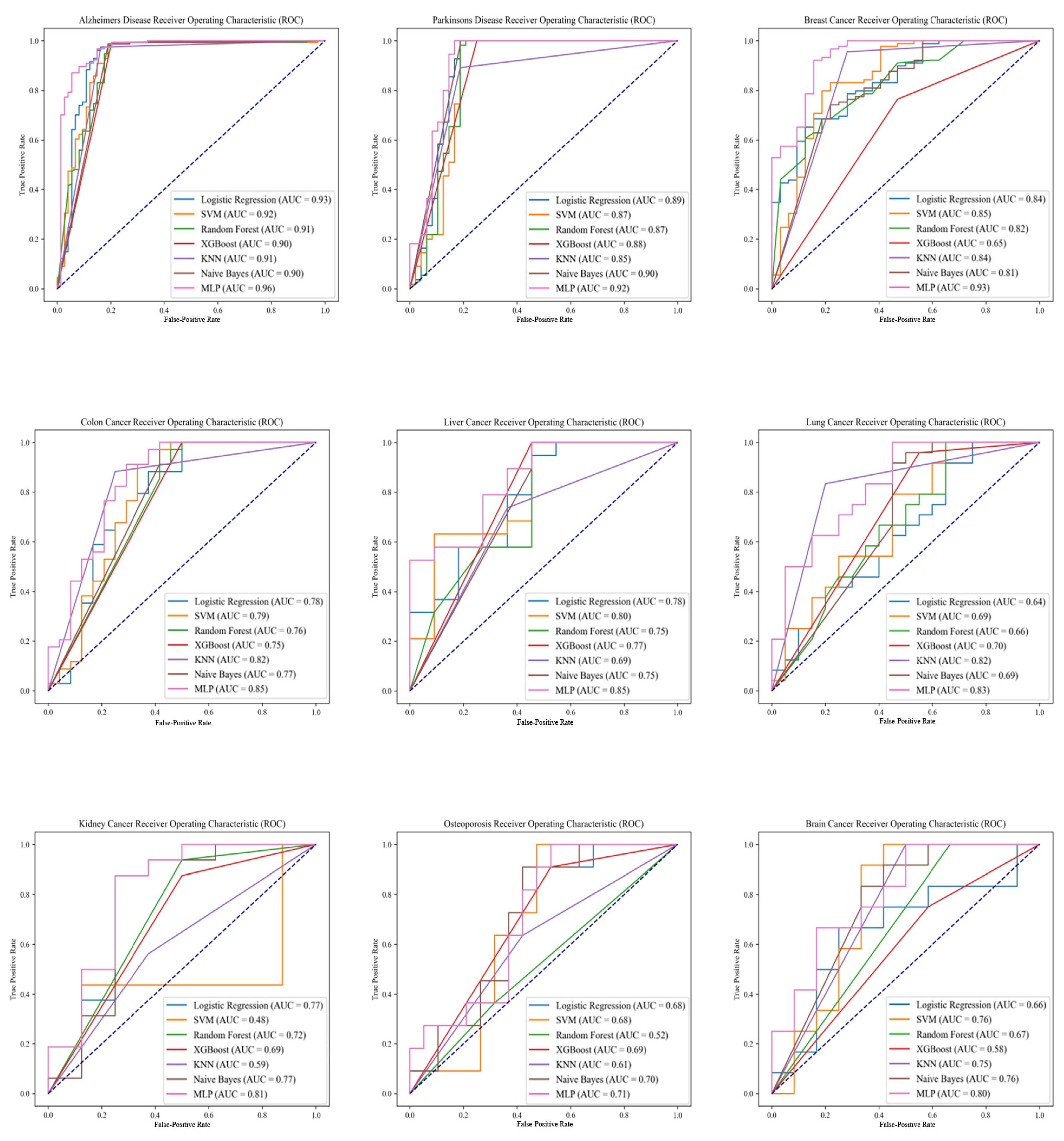
2.2. Model Performance Analysis
3. Discussions
3.1. Comparative Analysis of Machine Learning Models
3.2. Performance Variability among Different Diseases
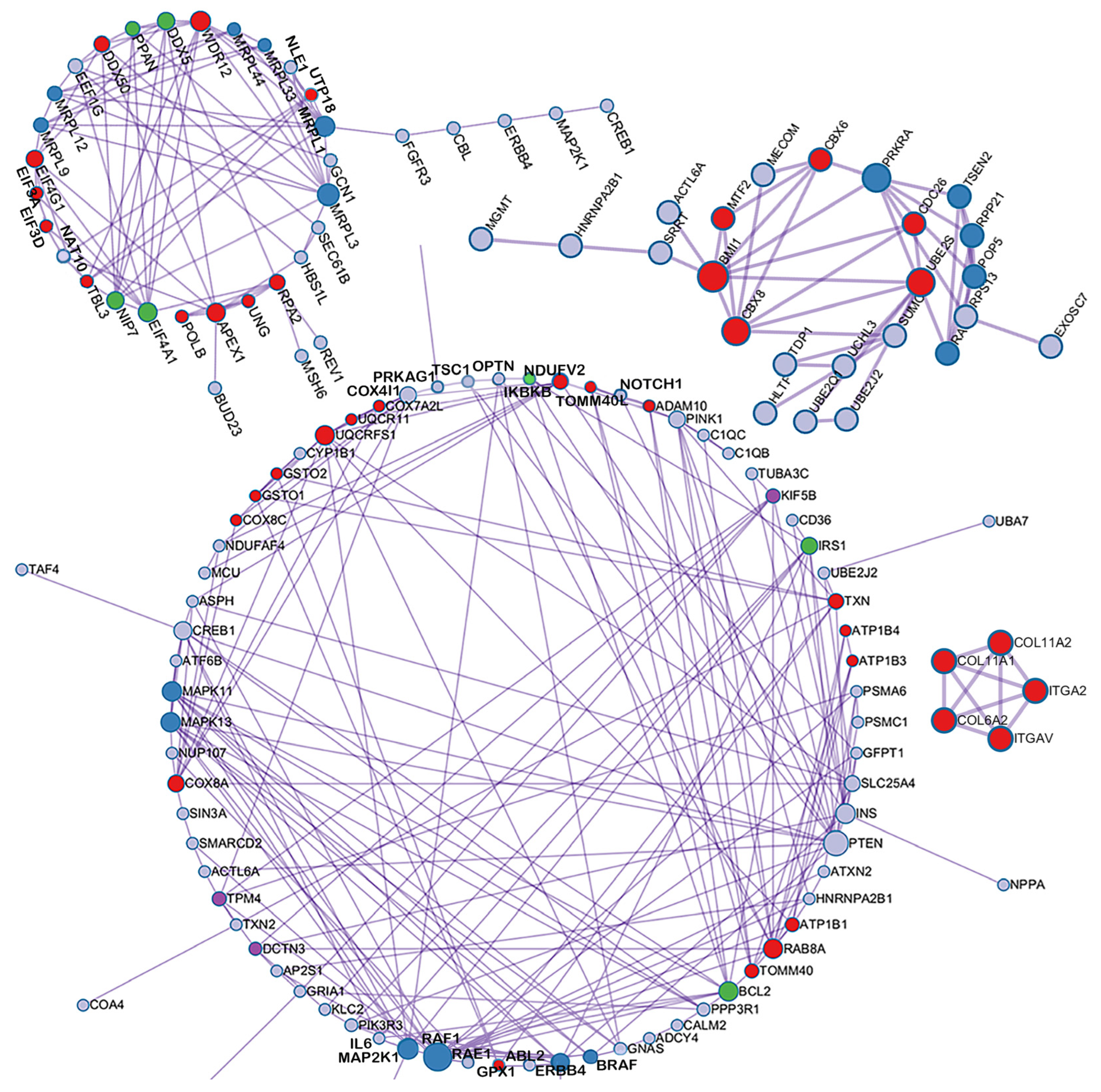
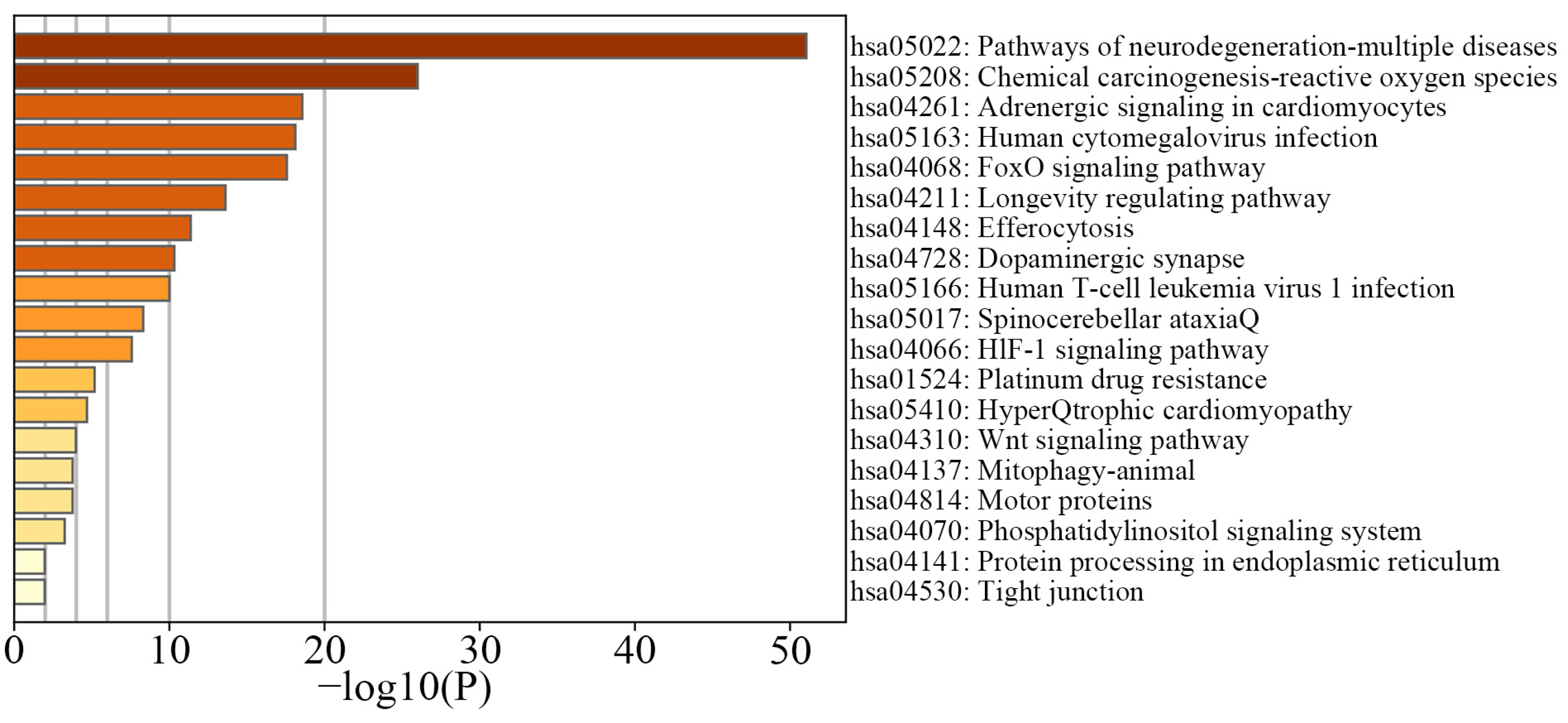
3.3. Insights from DNA Methylation Data Analysis
3.4. Limitations and Future Work
4. Materials and Methods
4.1. Datasets
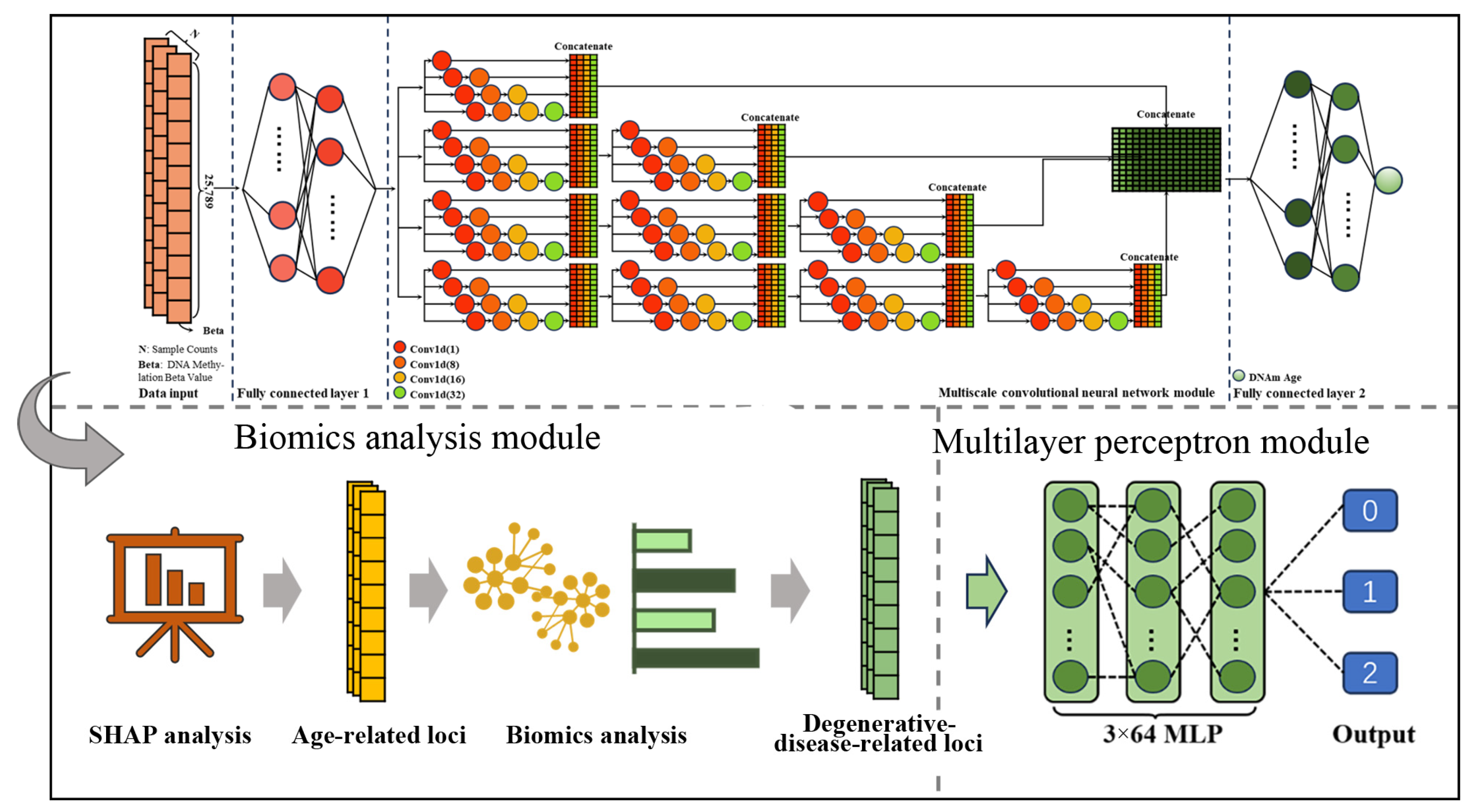
4.2. Model Construction
4.3. Epigenetic Clock Based on Deep Learning
4.4. Identification of Age-Related CpG Sites
4.5. Enrichment Analysis
4.6. Hyperparameter Tuning and Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chaudhary, M.R.; Chaudhary, S.; Sharma, Y.; Singh, T.A.; Mishra, A.K.; Sharma, S.; Mehdi, M.M. Aging, oxidative stress and degenerative diseases: Mechanisms, complications and emerging therapeutic strategies. Biogerontology 2023, 24, 609–662. [Google Scholar] [CrossRef]
- Self, W.K.; Holtzman, D.M. Emerging diagnostics and therapeutics for Alzheimer disease. Nat. Med. 2023, 29, 2187–2199. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.-W.W.; Shafieizadeh, K.; Sung, K.S.; Kwon, J. Influential factors toward the quality of dining services at continuing care retirement communities: Manager’s perspective. J. Hosp. Tour. Manag. 2023, 56, 60–68. [Google Scholar] [CrossRef]
- Simpson, D.J.; Chandra, T. Epigenetic age prediction. Aging Cell 2021, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Garcia-Closas, M.; Orr, N.; Fletcher, O.; Jones, M.; Ashworth, A.; Swerdlow, A.; Thorne, H.; Riboli, E.; Vineis, P. Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012, 72, 2304–2313. [Google Scholar] [CrossRef]
- van Veldhoven, K.; Polidoro, S.; Baglietto, L.; Severi, G.; Sacerdote, C.; Panico, S.; Mattiello, A.; Palli, D.; Masala, G.; Krogh, V. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clin. Epigenet. 2015, 7, 67. [Google Scholar] [CrossRef]
- Huang, M.; Xu, S.; Liu, L.; Zhang, M.; Guo, J.; Yuan, Y.; Xu, J.; Chen, X.; Zou, J. m6A methylation regulates osteoblastic differentiation and bone remodeling. Front. Cell Dev. Biol. 2021, 9, 783322. [Google Scholar] [CrossRef]
- Kenny, J.; Mullin, B.H.; Tomlinson, W.; Robertson, B.; Yuan, J.; Chen, W.; Zhao, J.; Pavlos, N.J.; Walsh, J.P.; Wilson, S.G. Age-dependent genetic regulation of osteoarthritis: Independent effects of immune system genes. Arthritis Res. Ther. 2023, 25, 232. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Vallerga, C.L.; Walker, R.M.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.S.; Fowdar, J.; Kennedy, M.; et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Hai, B.; Kuang, Z.; Wang, H.; Zhao, J. ResnetAge: A Resnet-Based DNA Methylation Age Prediction Method. Bioengineering 2023, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, H.; Qu, J.; Zong, X.; Liu, Y.; Kuang, Z.; Wang, H. A multi-organization epigenetic age prediction based on a channel attention perceptron networks. Front. Genet. 2024, 15, 1393856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Zhao, Y.; Liao, X.K.; Shi, W.Q.; Li, K.L.; Zou, Q.; Peng, S.L. Deep learning in omics: A survey and guideline. Brief. Funct. Genom. 2019, 18, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Yilmaz, A.; Saiyed, N.M.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Precision cardiovascular medicine: Artificial intelligence and epigenetics for the pathogenesis and prediction of coarctation in neonates. J. Matern.-Fetal Neonatal Med. 2022, 35, 457–464. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Er, A.; Aydas, B.; Turkoglu, O.; Taskin, B.D.; Duman, M.; Yilmaz, D.; Radhakrishna, U. Artificial intelligence and the detection of pediatric concussion using epigenomic analysis. Brain Res. 2020, 1726, 146510. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Xiao, Y.; Wu, J.; Zhao, X.; Li, H. A convolutional neural network based ensemble method for cancer prediction using DNA methylation data. In Proceedings of the 2019 11th International Conference on Machine Learning and Computing, Zhuhai China, 22–24 February 2019; pp. 191–196. [Google Scholar]
- Li, H. K-Nearest Neighbor. In Machine Learning Methods; Springer: Berlin/Heidelberg, Germany, 2023; pp. 55–66. [Google Scholar]
- Reddy, E.M.K.; Gurrala, A.; Hasitha, V.B.; Kumar, K.V.R. Introduction to Naive Bayes and a review on its subtypes with applications. In Bayesian Reasoning and Gaussian Processes for Machine Learning Applications; Chapman and Hall/CRC: Boca Raton, FL, USA, 2022; pp. 1–14. [Google Scholar]
- Rigatti, S.J. Random forest. J. Insur. Med. 2017, 47, 31–39. [Google Scholar] [CrossRef]
- Suthaharan, S. Machine Learning Models and Algorithms for Big Data Classification; Springer: Berlin/Heidelberg, Germany, 2016; Volume 36, pp. 1–12. [Google Scholar]
- Chen, T.; Guestrin, C. In Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Chicco, D. geneExpressionFromGEO: An R package to facilitate data reading from gene expression Omnibus (GEO). In Microarray Data Analysis; R Company: New York, NY, USA, 2022; pp. 187–194. [Google Scholar]
- Wang, H.; Cai, R.; Zong, X.; He, Z.; Zhang, L. MSCAP: DNA Methylation Age Predictor based on Multiscale Convolutional Neural Network. In Proceedings of the 2023 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Istanbul, Turkiye, 5–8 December 2023; IEEE: New York, NY, USA, 2023; pp. 3582–3586. [Google Scholar]
- Chen, Y.-a.; Choufani, S.; Ferreira, J.C.; Grafodatskaya, D.; Butcher, D.T.; Weksberg, R. Sequence overlap between autosomal and sex-linked probes on the Illumina HumanMethylation27 microarray. Genomics 2011, 97, 214–222. [Google Scholar] [CrossRef]
- Vanderlinden, L.A.; Johnson, R.K.; Carry, P.M.; Dong, F.; DeMeo, D.L.; Yang, I.V.; Norris, J.M.; Kechris, K. An effective processing pipeline for harmonizing DNA methylation data from Illumina’s 450K and EPIC platforms for epidemiological studies. BMC Res. Notes 2021, 14, 352. [Google Scholar] [CrossRef]
- Chen, H.; Lundberg, S.M.; Lee, S.I. Explaining a series of models by propagating Shapley values. Nat. Commun. 2022, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeck, G.; Lykov, A.; Schleich, M.; Suciu, D. On the Tractability of SHAP Explanations. J. Artif. Intell. Res. 2022, 74, 851–886. [Google Scholar] [CrossRef]
- Marcílio, W.E.; Eler, D.M. From explanations to feature selection: Assessing SHAP values as feature selection mechanism. In Proceedings of the 2020 33rd SIBGRAPI conference on Graphics, Patterns and Images (SIBGRAPI), Porto de Galinhas, Brazil, 7–10 November 2020; IEEE: New York, NY, USA, 2020; pp. 340–347. [Google Scholar]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.T.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2020, 49, D884–D891. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Legeay, M.; Doncheva, N.T.; Morris, J.H.; Jensen, L.J. Visualize omics data on networks with Omics Visualizer, a Cytoscape App. F1000Research 2020, 9, 157. [Google Scholar] [CrossRef]
- Jansen, I.E.; van der Lee, S.J.; Gomez-Fonseca, D.; De Rojas, I.; Dalmasso, M.C.; Grenier-Boley, B.; Zettergren, A.; Mishra, A.; Ali, M.; Andrade, V. Genome-wide meta-analysis for Alzheimer’s disease cerebrospinal fluid biomarkers. Acta Neuropathol. 2022, 144, 821–842. [Google Scholar] [CrossRef]
- Juan, H.F.; Lin, J.Y.C.; Chang, W.H.; Wu, C.Y.; Pan, T.L.; Tseng, M.J.; Khoo, K.H.; Chen, S.T. Biomic study of human myeloid leukemia cells differentiation to macrophages using DNA array, proteomic, and bioinformatic analytical methods. Electrophoresis 2002, 23, 2490–2504. [Google Scholar] [CrossRef]
- Huang, F.; Fu, M.; Li, J.; Chen, L.; Feng, K.; Huang, T.; Cai, Y.-D. Analysis and prediction of protein stability based on interaction network, gene ontology, and KEGG pathway enrichment scores. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2023, 1871, 140889. [Google Scholar] [CrossRef] [PubMed]
- Safari-Alighiarloo, N.; Taghizadeh, M.; Rezaei-Tavirani, M.; Goliaei, B.; Peyvandi, A.A. Protein-protein interaction networks (PPI) and complex diseases. Gastroenterol. Hepatol. Bed Bench 2014, 7, 17. [Google Scholar] [PubMed]
- Wang, S.; Wu, R.X.; Lu, J.Q.; Jiang, Y.J.; Huang, T.; Cai, Y.D. Protein-protein interaction networks as miners of biological discovery. Proteomics 2022, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 10. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. biorxiv 2016, 060012. [Google Scholar] [CrossRef]
- Tipney, H.; Hunter, L. An introduction to effective use of enrichment analysis software. Hum. Genom. 2010, 4, 202–206. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Degenerative Disease Type | AUC | ACC | Recall | Precision |
|---|---|---|---|---|
| Alzheimer’s Disease | 0.96 | 0.92 | 0.97 | 0.92 |
| Parkinson’s Disease | 0.92 | 0.90 | 0.96 | 0.87 |
| Breast Cancer | 0.93 | 0.91 | 0.97 | 0.91 |
| Colon Cancer | 0.85 | 0.83 | 0.97 | 0.79 |
| Lung Cancer | 0.85 | 0.77 | 0.89 | 0.77 |
| Liver Cancer | 0.83 | 0.73 | 0.62 | 0.83 |
| Kidney Cancer | 0.81 | 0.83 | 0.94 | 0.83 |
| Osteoporosis | 0.71 | 0.63 | 0.82 | 0.50 |
| Brain Cancer | 0.80 | 0.71 | 1.00 | 0.63 |
| (a) | |||||
| Disease Type | Model | AUC | ACC | Precision | Recall |
| Alzheimer’s Disease | MLP | 0.96 | 0.92 | 0.97 | 0.92 |
| LR | 0.93 | 0.93 | 0.99 | 0.91 | |
| SVM | 0.92 | 0.93 | 0.99 | 0.91 | |
| RF | 0.91 | 0.93 | 0.99 | 0.91 | |
| XGBoost | 0.90 | 0.93 | 0.99 | 0.91 | |
| KNN | 0.91 | 0.93 | 0.97 | 0.93 | |
| NB | 0.90 | 0.93 | 0.98 | 0.92 | |
| Parkinson’s Disease | MLP | 0.92 | 0.90 | 0.96 | 0.87 |
| LR | 0.89 | 0.90 | 0.98 | 0.86 | |
| SVM | 0.87 | 0.91 | 1.00 | 0.86 | |
| RF | 0.87 | 0.90 | 0.98 | 0.86 | |
| XGBoost | 0.88 | 0.88 | 1.00 | 0.82 | |
| KNN | 0.85 | 0.85 | 0.89 | 0.84 | |
| NB | 0.90 | 0.91 | 1.00 | 0.86 | |
| Breast Cancer | MLP | 0.93 | 0.91 | 0.97 | 0.91 |
| LR | 0.84 | 0.79 | 1.00 | 0.77 | |
| SVM | 0.85 | 0.86 | 1.00 | 0.84 | |
| RF | 0.82 | 0.74 | 0.79 | 0.85 | |
| (b) | |||||
| Disease Type | Model | AUC | ACC | Precision | Recall |
| Breast Cancer | XGBoost | 0.65 | 0.70 | 0.76 | 0.82 |
| KNN | 0.84 | 0.89 | 0.96 | 0.90 | |
| NB | 0.81 | 0.72 | 0.69 | 0.91 | |
| Colon Cancer | MLP | 0.85 | 0.83 | 0.97 | 0.79 |
| LR | 0.78 | 0.59 | 1.00 | 0.59 | |
| SVM | 0.79 | 0.59 | 1.00 | 0.59 | |
| RF | 0.76 | 0.79 | 1.00 | 0.74 | |
| XGBoost | 0.75 | 0.79 | 1.00 | 0.74 | |
| KNN | 0.82 | 0.83 | 0.88 | 0.83 | |
| NB | 0.77 | 0.78 | 0.91 | 0.76 | |
| Lung Cancer | MLP | 0.85 | 0.77 | 0.89 | 0.77 |
| LR | 0.78 | 0.63 | 1.00 | 0.63 | |
| SVM | 0.80 | 0.77 | 0.89 | 0.77 | |
| RF | 0.75 | 0.83 | 1.00 | 0.79 | |
| XGBoost | 0.77 | 0.83 | 1.00 | 0.79 | |
| KNN | 0.69 | 0.70 | 0.74 | 0.78 | |
| NB | 0.75 | 0.83 | 1.00 | 0.79 | |
| Liver Cancer | MLP | 0.83 | 0.73 | 0.62 | 0.83 |
| LR | 0.64 | 0.55 | 1.00 | 0.55 | |
| SVM | 0.69 | 0.64 | 0.79 | 0.63 | |
| RF | 0.66 | 0.61 | 0.71 | 0.63 | |
| XGBoost | 0.70 | 0.73 | 0.96 | 0.68 | |
| KNN | 0.82 | 0.82 | 0.83 | 0.83 | |
| NB | 0.69 | 0.70 | 0.83 | 0.69 | |
| Kidney Cancer | MLP | 0.81 | 0.83 | 0.94 | 0.83 |
| LR | 0.77 | 0.67 | 1.00 | 0.67 | |
| SVM | 0.48 | 0.67 | 1.00 | 0.67 | |
| RF | 0.72 | 0.79 | 0.94 | 0.79 | |
| XGBoost | 0.69 | 0.75 | 0.88 | 0.78 | |
| KNN | 0.59 | 0.58 | 0.56 | 0.75 | |
| NB | 0.77 | 0.79 | 0.94 | 0.79 | |
| Osteoporosis | MLP | 0.71 | 0.63 | 0.82 | 0.50 |
| LR | 0.68 | 0.37 | 1.00 | 0.37 | |
| SVM | 0.68 | 0.37 | 1.00 | 0.37 | |
| RF | 0.52 | 0.57 | 0.36 | 0.40 | |
| XGBoost | 0.69 | 0.63 | 0.91 | 0.50 | |
| KNN | 0.61 | 0.60 | 0.64 | 0.47 | |
| NB | 0.70 | 0.60 | 1.00 | 0.48 | |
| (c) | |||||
| Disease Type | Model | AUC | ACC | Precision | Recall |
| Brain Cancer | MLP | 0.80 | 0.71 | 1.00 | 0.63 |
| LR | 0.66 | 0.50 | 1.00 | 0.50 | |
| SVM | 0.76 | 0.50 | 1.00 | 0.50 | |
| RF | 0.67 | 0.67 | 1.00 | 0.60 | |
| XGBoost | 0.58 | 0.58 | 0.75 | 0.56 | |
| KNN | 0.75 | 0.75 | 1.00 | 0.67 | |
| NB | 0.76 | 0.75 | 0.83 | 0.71 | |
| Disease | Stage | Sample Count |
|---|---|---|
| Alzheimer’s Disease | Early | 401 |
| Late | 321 | |
| Parkinson’s Disease | Early | 229 |
| Late | 238 | |
| Breast Cancer | Early | 148 |
| Late | 134 | |
| Colon Cancer | Early | 146 |
| Late | 121 | |
| Lung Cancer | Early | 84 |
| Late | 104 | |
| Liver Cancer | Early | 68 |
| Late | 46 | |
| Kidney Cancer | Early | 37 |
| Late | 39 | |
| Osteoporosis | Early | 38 |
| Late | 34 | |
| Brain Cancer | Early | 33 |
| Late | 27 |
| CpG ID | SHAP Values | Gene Name | Chromosome Position |
|---|---|---|---|
| cg00008493 | −3.31 × 10−5 | COX8C | 14 |
| cg00024812 | −5.67 × 10−5 | CPSF3 | 2 |
| cg00033773 | −4.54 × 10−5 | MORG1 | 19 |
| cg00094319 | −2.96 × 10−5 | PABPC3 | 13 |
| cg00103783 | −8.55 × 10−5 | MPDU1 | 17 |
| cg00136477 | −7.97 × 10−5 | C1QC | 1 |
| cg00187686 | −2.08 × 10−5 | TCN1 | 11 |
| cg00234961 | −2.22 × 10−5 | ZBED4 | 22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Cai, R.; Wang, C.; Liu, J.; Kuang, Z.; Wang, H. Prediction of Multiple Degenerative Diseases Based on DNA Methylation in a Co-Physiology Mechanisms Perspective. Int. J. Mol. Sci. 2024, 25, 9514. https://doi.org/10.3390/ijms25179514
Zhang L, Cai R, Wang C, Liu J, Kuang Z, Wang H. Prediction of Multiple Degenerative Diseases Based on DNA Methylation in a Co-Physiology Mechanisms Perspective. International Journal of Molecular Sciences. 2024; 25(17):9514. https://doi.org/10.3390/ijms25179514
Chicago/Turabian StyleZhang, Li, Ruirui Cai, Chencai Wang, Jialong Liu, Zhejun Kuang, and Han Wang. 2024. "Prediction of Multiple Degenerative Diseases Based on DNA Methylation in a Co-Physiology Mechanisms Perspective" International Journal of Molecular Sciences 25, no. 17: 9514. https://doi.org/10.3390/ijms25179514