
Mitochondrial NME6 Influences Basic Cellular Processes in Tumor Cells In Vitro




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
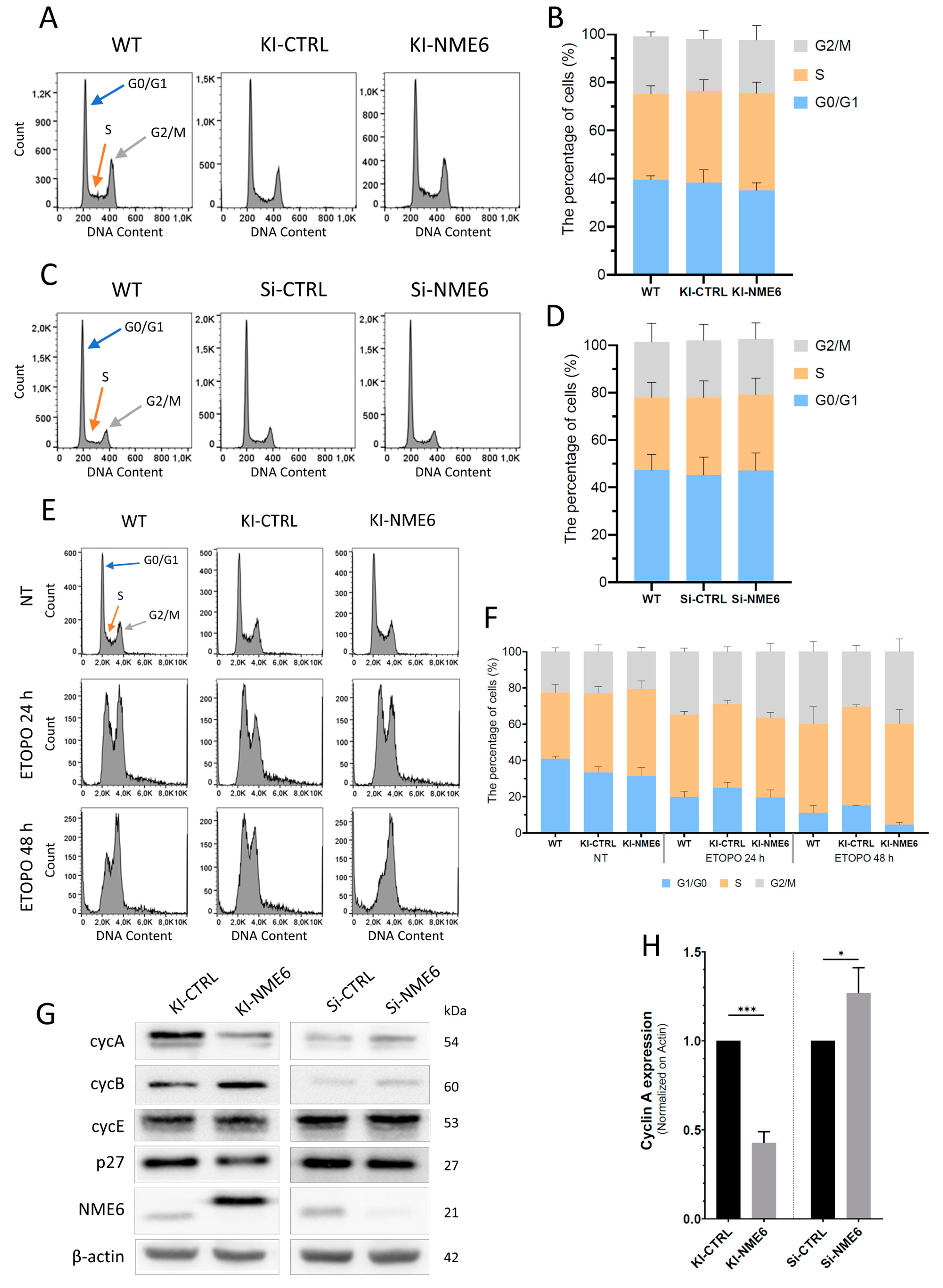
2.1. NME6 Overexpression Influences Cell Cycle Progression after DNA Damage and Changes the Level of Cell Cycle Regulators
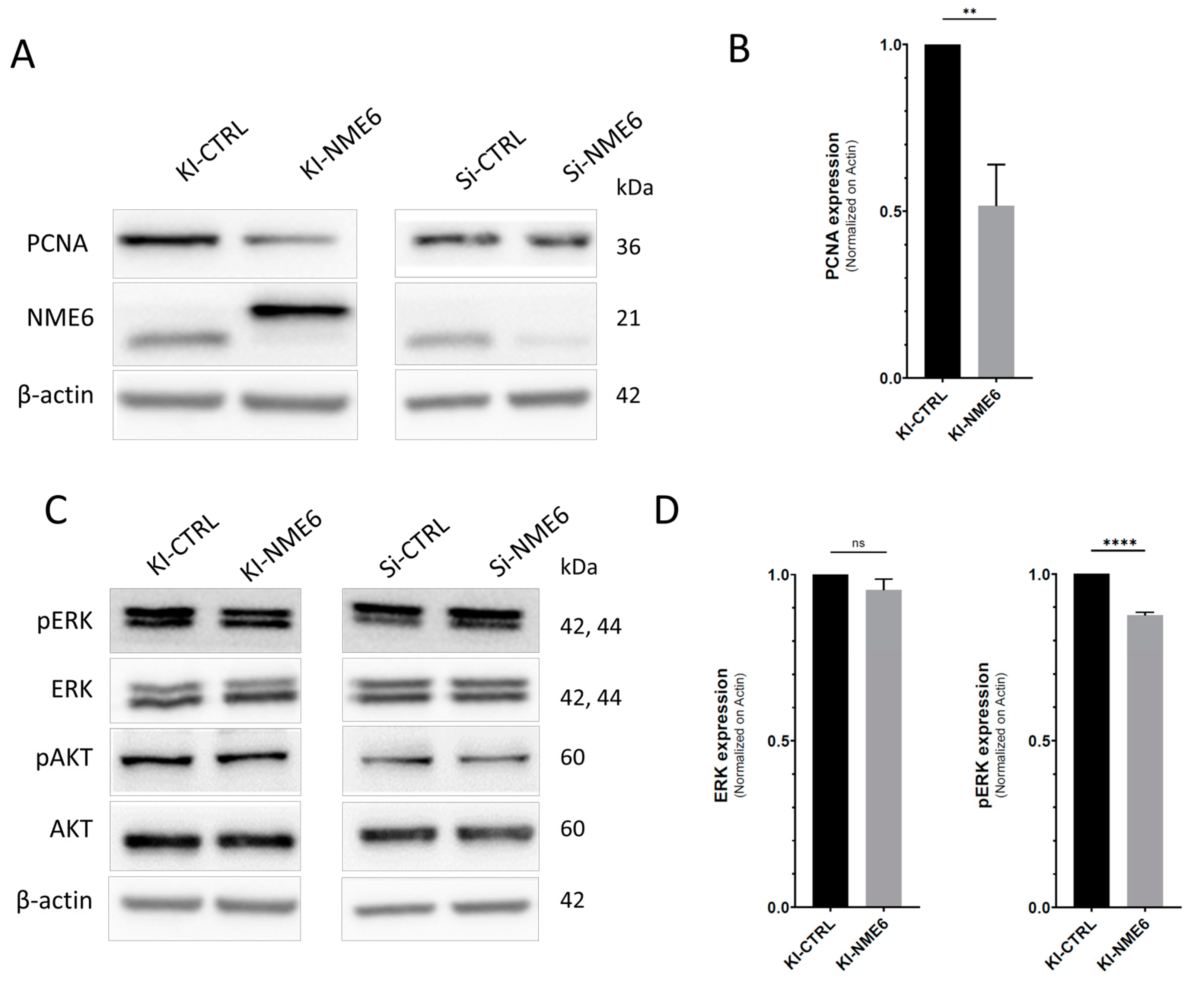
2.2. NME6 Overexpression Reduces Levels of PCNA Proliferation Marker and Moderately Affects MAPK/ERK Pathway Activity
2.3. NME6 Overexpression Decreases Migration and Alters the Expression of EMT Markers
2.4. NME6 Is Not Associated with Apoptosis but Moderately Affects the Level of p53 Family Members in Unstressed Conditions
3. Materials and Methods
3.1. Cells and Cell Maintenance
3.2. Cell Cycle
3.3. Apoptosis
3.4. Wound-Healing Assay
3.5. Protein Extraction and Western Blotting
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lascu, I.; Gonin, P. The Catalytic Mechanism of Nucleoside Diphosphate Kinases. J. Bioenerg. Biomembr. 2000, 32, 237–246. [Google Scholar] [CrossRef]
- Lacombe, M.L.; Milon, L.; Munier, A.; Mehus, J.G.; Lambeth, D.O. The Human Nm23/Nucleoside Diphosphate Kinases. J. Bioenerg. Biomembr. 2000, 32, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, T.; Pontarotti, P.; Fauvel, C.; Bobe, J. Nme Protein Family Evolutionary History, a Vertebrate Perspective. BMC Evol. Biol. 2009, 9, 256. [Google Scholar] [CrossRef]
- Gilles, A.M.; Presecan, E.; Vonica, A.; Lascu, I. Nucleoside Diphosphate Kinase from Human Erythrocytes. Structural Characterization of the Two Polypeptide Chains Responsible for Heterogeneity of the Hexameric Enzyme. J. Biol. Chem. 1991, 266, 8784–8789. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S.; Bevilacqua, G.; Kopper, L.; Thorgeirsson, U.P.; Talmadge, J.E.; Liotta, L.A.; Sobel, M.E. Evidence for a Novel Gene Associated With Low Tumor Metastatic Potential. JNCI J. Natl. Cancer Inst. 1988, 80, 200–204. [Google Scholar] [CrossRef]
- Ćetković, H.; Harcet, M.; Roller, M.; Herak Bosnar, M. A Survey of Metastasis Suppressors in Metazoa. Lab. Investig. 2018, 98, 554–570. [Google Scholar] [CrossRef] [PubMed]
- Herak Bosnar, M.; Dubravčić, K.; Bago, R.; Pavelić, J. Head and Neck Tumor Cells Exhibit Altered Proliferation upon Overexpression of Nm23 Genes. Croat. Chem. Acta 2008, 81, 183–189. [Google Scholar]
- Ćetković, H.; Perina, D.; Harcet, M.; Mikoč, A.; Herak Bosnar, M. Nme Family of Proteins—Clues from Simple Animals. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 133–142. [Google Scholar] [CrossRef]
- Zhou, X.-B.; Feng, Y.-X.; Sun, Q.; Lukowski, R.; Qiu, Y.; Spiger, K.; Li, Z.; Ruth, P.; Korth, M.; Skolnik, E.Y.; et al. Nucleoside Diphosphate Kinase B–Activated Intermediate Conductance Potassium Channels Are Critical for Neointima Formation in Mouse Carotid Arteries. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1852–1861. [Google Scholar] [CrossRef]
- Cipollini, G.; Berti, A.; Fiore, L.; Rainaldi, G.; Basolo, F.; Merlo, G.; Bevilacqua, G.; Caligo, M.A. Down-Regulation of the Nm23.H1 Gene Inhibits Cell Proliferation. Int. J. Cancer 1997, 73, 297–302. [Google Scholar] [CrossRef]
- Lombardi, D.; Lacombe, M.-L.; Paggi, M.G. Nm23: Unraveling Its Biological Function in Cell Differentiation. J. Cell. Physiol. 2000, 182, 144–149. [Google Scholar] [CrossRef]
- Bilitou, A.; Watson, J.; Gartner, A.; Ohnuma, S. The NM23 Family in Development. Mol. Cell. Biochem. 2009, 329, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Lakso, M.; Steeg, P.S.; Westphal, H. Embryonic Expression of Nm23 during Mouse Organogenesis. Cell Growth Differ. 1992, 3, 873–879. [Google Scholar]
- Boissan, M.; Montagnac, G.; Shen, Q.; Griparic, L.; Guitton, J.; Romao, M.; Sauvonnet, N.; Lagache, T.; Lascu, I.; Raposo, G.; et al. Nucleoside Diphosphate Kinases Fuel Dynamin Superfamily Proteins with GTP for Membrane Remodeling. Science 2014, 344, 1510–1515. [Google Scholar] [CrossRef]
- Fournier, H.-N.; Albigès-Rizo, C.; Block, M.R. New Insights into Nm23 Control of Cell Adhesion and Migration. J. Bioenerg. Biomembr. 2003, 35, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tong, Y.; Wong, Y.H. Regulatory Functions of Nm23-H2 in Tumorigenesis: Insights from Biochemical to Clinical Perspectives. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 243–256. [Google Scholar] [CrossRef]
- Feng, Y.; Gross, S.; Wolf, N.M.; Butenschön, V.M.; Qiu, Y.; Devraj, K.; Liebner, S.; Kroll, J.; Skolnik, E.Y.; Hammes, H.-P.; et al. Nucleoside Diphosphate Kinase B Regulates Angiogenesis Through Modulation of Vascular Endothelial Growth Factor Receptor Type 2 and Endothelial Adherens Junction Proteins. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2292–2300. [Google Scholar] [CrossRef]
- Chen, C.-W.; Wang, H.-L.; Huang, C.-W.; Huang, C.-Y.; Lim, W.K.; Tu, I.-C.; Koorapati, A.; Hsieh, S.-T.; Kan, H.-W.; Tzeng, S.-R.; et al. Two Separate Functions of NME3 Critical for Cell Survival Underlie a Neurodegenerative Disorder. Proc. Natl. Acad. Sci. USA 2019, 116, 566–574. [Google Scholar] [CrossRef]
- Milon, L.; Rousseau-Merck, M.F.; Munier, A.; Erent, M.; Lascu, L.; Capeau, J.; Lacombe, M.L. Nm23-H4, a New Member of the Family of Human Nm23/Nucleoside Diphosphate Kinase Genes Localised on Chromosome 16p13. Hum. Genet. 1997, 99, 550–557. [Google Scholar] [CrossRef]
- Desvignes, T.; Pontarotti, P.; Bobe, J. Nme Gene Family Evolutionary History Reveals Pre-Metazoan Origins and High Conservation between Humans and the Sea Anemone, Nematostella Vectensis. PLoS ONE 2010, 5, e15506. [Google Scholar] [CrossRef]
- Mehus, J.G.; Deloukas, P.; Lambeth, D.O. NME6: A New Member of the Nm23 /Nucleoside Diphosphate Kinase Gene Family Located on Human Chromosome 3p21.3. Hum. Genet. 1999, 104, 454–459. [Google Scholar] [CrossRef]
- Proust, B.L.J. Human NME6 Protein: Subcellular Localization, Structure and Function. Ph.D. Thesis, University of Zagreb, Zagreb, Croatia, 2022. [Google Scholar]
- Boissan, M.; Schlattner, U.; Lacombe, M.-L. The NDPK/NME Superfamily: State of the Art. Lab. Investig. 2018, 98, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Herak Bosnar, M.; Radić, M.; Ćetković, H. A Young Researcher’s Guide to NME/Nm23/NDP Kinase. Period. Biol. 2018, 120, 3–9. [Google Scholar] [CrossRef]
- Tsuiki, H.; Nitta, M.; Furuya, A.; Hanai, N.; Fujiwara, T.; Inagaki, M.; Kochi, M.; Ushio, Y.; Saya, H.; Nakamura, H. A Novel Human Nucleoside Diphosphate (NDP) Kinase, Nm23-H6, Localizes in Mitochondria and Affects Cytokinesis. J. Cell. Biochem. 2000, 76, 254–269. [Google Scholar] [CrossRef]
- Proust, B.; Radić, M.; Vidaček, N.Š.; Cottet, C.; Attia, S.; Lamarche, F.; Ačkar, L.; Mikulčić, V.G.; Tokarska-Schlattner, M.; Ćetković, H.; et al. NME6 Is a Phosphotransfer-Inactive, Monomeric NME/NDPK Family Member and Functions in Complexes at the Interface of Mitochondrial Inner Membrane and Matrix. Cell Biosci. 2021, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Grotehans, N.; McGarry, L.; Nolte, H.; Xavier, V.; Kroker, M.; Narbona-Pérez, Á.J.; Deshwal, S.; Giavalisco, P.; Langer, T.; MacVicar, T. Ribonucleotide Synthesis by NME6 Fuels Mitochondrial Gene Expression. EMBO J. 2023, 42, e113256. [Google Scholar] [CrossRef]
- Kramer, N.J.; Prakash, G.; Isaac, R.S.; Choquet, K.; Soto, I.; Petrova, B.; Merens, H.E.; Kanarek, N.; Churchman, L.S. Regulators of Mitonuclear Balance Link Mitochondrial Metabolism to MtDNA Expression. Nat. Cell Biol. 2023, 25, 1575–1589. [Google Scholar] [CrossRef]
- Seifert, M.; Welter, C.; Mehraein, Y.; Seitz, G. Expression of the Nm23 Homologues Nm23-H4, Nm23-H6, and Nm23-H7 in Human Gastric and Colon Cancer. J. Pathol. 2005, 205, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Lou, J.; Zhong, R.; Chen, X.; Li, J.; Liu, C.; Gong, Y.; Yang, Y.; Zhu, Y.; Zhang, Y.; et al. Identification of a Potential Regulatory Variant for Colorectal Cancer Risk Mapping to 3p21.31 in Chinese Population. Sci. Rep. 2016, 6, 25194. [Google Scholar] [CrossRef]
- Wang, C.-H.; Ma, N.; Lin, Y.-T.; Wu, C.-C.; Hsiao, M.; Lu, F.L.; Yu, C.-C.; Chen, S.-Y.; Lu, J. A ShRNA Functional Screen Reveals Nme6 and Nme7 Are Crucial for Embryonic Stem Cell Renewal. Stem Cells 2012, 30, 2199–2211. [Google Scholar] [CrossRef]
- Ernst, O.; Sun, J.; Lin, B.; Banoth, B.; Dorrington, M.G.; Liang, J.; Schwarz, B.; Stromberg, K.A.; Katz, S.; Vayttaden, S.J.; et al. A Genome-Wide Screen Uncovers Multiple Roles for Mitochondrial Nucleoside Diphosphate Kinase D in Inflammasome Activation. Sci. Signal. 2021, 14, eabe0387. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Lossaint, G.; Horvat, A.; Gire, V.; Bačević, K.; Mrouj, K.; Charrier-Savournin, F.; Georget, V.; Fisher, D.; Dulić, V. Reciprocal Regulation of P21 and Chk1 Controls the Cyclin D1-RB Pathway to Mediate Senescence Onset after G2 Arrest. J. Cell Sci. 2022, 135, jcs259114. [Google Scholar] [CrossRef] [PubMed]
- Diller, L.; Kassel, J.; Nelson, C.E.; Gryka, M.A.; Litwak, G.; Gebhardt, M.; Bressac, B.; Ozturk, M.; Baker, S.J.; Vogelstein, B.; et al. P53 Functions as a Cell Cycle Control Protein in Osteosarcomas. Mol. Cell. Biol. 1990, 10, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Pruteanu, L.L.; Braicu, C.; Módos, D.; Jurj, M.A.; Raduly, L.Z.; Zănoagă, O.; Magdo, L.; Cojocneanu, R.; Paşca, S.; Moldovan, C.; et al. Targeting Cell Death Mechanism Specifically in Triple Negative Breast Cancer Cell Lines. Int. J. Mol. Sci. 2022, 23, 4784. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Roa, M.; Malumbres, M. Fueling the Cell Division Cycle. Trends Cell Biol. 2017, 27, 69–81. [Google Scholar] [CrossRef]
- Sakamaki, T.; Casimiro, M.C.; Ju, X.; Quong, A.A.; Katiyar, S.; Liu, M.; Jiao, X.; Li, A.; Zhang, X.; Lu, Y.; et al. Cyclin D1 Determines Mitochondrial Function InVivo. Mol. Cell. Biol. 2006, 26, 5449–5469. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-Related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A Hyperfused Mitochondrial State Achieved at G1-S Regulates Cyclin E Buildup and Entry into S Phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef]
- Wang, Z.; Fan, M.; Candas, D.; Zhang, T.Q.; Qin, L.; Eldridge, A.; Wachsmann-Hogiu, S.; Ahmed, K.M.; Chromy, B.A.; Nantajit, D.; et al. Cyclin B1/Cdk1 Coordinates Mitochondrial Respiration for Cell-Cycle G2/M Progression. Dev. Cell 2014, 29, 217–232. [Google Scholar] [CrossRef]
- Rosenthal, C.K. Cdk1 Boosts Mitochondrial Energy Production. Nat. Cell Biol. 2014, 16, 511. [Google Scholar] [CrossRef]
- Poikonen, P.; Sjöström, J.; Amini, R.M.; Villman, K.; Ahlgren, J.; Blomqvist, C. Cyclin A as a Marker for Prognosis and Chemotherapy Response in Advanced Breast Cancer. Br. J. Cancer 2005, 93, 515–519. [Google Scholar] [CrossRef]
- Dutta, A.; Chandra, R.; Leiter, L.M.; Lester, S. Cyclins as Markers of Tumor Proliferation: Immunocytochemical Studies in Breast Cancer. Proc. Natl. Acad. Sci. USA 1995, 92, 5386–5390. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Liu, T.; Liu, Q.; Yang, L.; Zhang, Q.; Han, X.; Shen, T.; Zhang, X.; Lu, X. Widely Targeted Metabolomic Analyses Unveil the Metabolic Variations after Stable Knock-down of NME4 in Esophageal Squamous Cell Carcinoma Cells. Mol. Cell. Biochem. 2020, 471, 81–89. [Google Scholar] [CrossRef]
- Wang, W.; Dong, M.; Cui, J.; Xu, F.; Yan, C.; Ma, C.; Yi, L.; Tang, W.; Dong, J.; Wei, Y. NME4 May Enhance Non-small Cell Lung Cancer Progression by Overcoming Cell Cycle Arrest and Promoting Cellular Proliferation. Mol. Med. Rep. 2019, 20, 1629–1636. [Google Scholar] [CrossRef]
- Zheng, S.; Liu, Q.; Liu, T.; Yang, L.; Zhang, Q.; Shen, T.; Zhang, X.; Han, X.; Lu, X. NME4 Modulates PD-L1 Expression via the STAT3 Signaling Pathway in Squamous Cell Carcinoma. Biochem. Biophys. Res. Commun. 2020, 526, 29–34. [Google Scholar] [CrossRef]
- Olaisen, C.; Müller, R.; Nedal, A.; Otterlei, M. PCNA-Interacting Peptides Reduce Akt Phosphorylation and TLR-Mediated Cytokine Secretion Suggesting a Role of PCNA in Cellular Signaling. Cell. Signal. 2015, 27, 1478–1487. [Google Scholar] [CrossRef]
- Paplomata, E.; O’regan, R. The PI3K/AKT/MTOR Pathway in Breast Cancer: Targets, Trials and Biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.; Bergin, O.; Bianchi, A.; McNally, S.; Martin, F. Key Signalling Nodes in Mammary Gland Development and Cancer. Mitogen-Activated Protein Kinase Signalling in Experimental Models of Breast Cancer Progression and in Mammary Gland Development. Breast Cancer Res. 2009, 11, 209. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Buján, J.; García-Honduvilla, N.; Coca, S. Signal Transduction Pathways in Breast Cancer: The Important Role of PI3K/Akt/MTOR. J. Oncol. 2020, 2020, 9258396. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Z.; Lu, N. A New Role for the PI3K/Akt Signaling Pathway in the Epithelial-Mesenchymal Transition. Cell Adhes. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Olea-Flores, M.; Zuñiga-Eulogio, M.D.; Mendoza-Catalán, M.A.; Rodríguez-Ruiz, H.A.; Castañeda-Saucedo, E.; Ortuño-Pineda, C.; Padilla-Benavides, T.; Navarro-Tito, N. Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial–Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 2885. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, M.-L.; Lamarche, F.; De Wever, O.; Padilla-Benavides, T.; Carlson, A.; Khan, I.; Huna, A.; Vacher, S.; Calmel, C.; Desbourdes, C.; et al. The Mitochondrially-Localized Nucleoside Diphosphate Kinase D (NME4) Is a Novel Metastasis Suppressor. BMC Biol. 2021, 19, 228. [Google Scholar] [CrossRef]
- Jonkman, J.E.N.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. Cell Adhesion & Migration An Introduction to the Wound Healing Assay Using Livecell Microscopy An Introduction to the Wound Healing Assay Using Livecell Microscopy. Cell Adhes. Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef]
- Zanotelli, M.R.; Goldblatt, Z.E.; Miller, J.P.; Bordeleau, F.; Li, J.; VanderBurgh, J.A.; Lampi, M.C.; King, M.R.; Reinhart-King, C.A. Regulation of ATP Utilization during Metastatic Cell Migration by Collagen Architecture. Mol. Biol. Cell 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Haerinck, J.; Berx, G. Partial EMT Takes the Lead in Cancer Metastasis. Dev. Cell 2021, 56, 3174–3176. [Google Scholar] [CrossRef]
- Bae, Y.-H.; Shin, J.-M.; Park, H.-J.; Jang, H.-O.; Bae, M.-K.; Bae, S.-K. Gain-of-Function Mutant P53-R280K Mediates Survival of Breast Cancer Cells. Genes Genom. 2014, 36, 171–178. [Google Scholar] [CrossRef]
- Moulder, D.E.; Hatoum, D.; Tay, E.; Lin, Y.; McGowan, E.M. The Roles of P53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer? Cancers 2018, 10, 189. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.-G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. P53 Regulates Mitochondrial Respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-Associated Mutant P53 Drives the Warburg Effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A Pivotal Role for P53: Balancing Aerobic Respiration and Glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef]
- Compton, S.; Kim, C.; Griner, N.B.; Potluri, P.; Scheffler, I.E.; Sen, S.; Jerry, D.J.; Schneider, S.; Yadava, N. Mitochondrial Dysfunction Impairs Tumor Suppressor P53 Expression/Function. J. Biol. Chem. 2011, 286, 20297–20312. [Google Scholar] [CrossRef]
- Buckley, N.; Craxton, A.; Sun, X.-M.; Panatta, E.; Pinon, L.; Llodrá, J.; Morone, N.; Amelio, I.; Melino, G.; Martins, L.M.; et al. TAp73 Regulates Mitochondrial Dynamics and Multiciliated Cell Homeostasis through an OPA1 Axis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Li, Y.; Yao, L.; Mori, Y.; Sun, S.X. On the Energy Efficiency of Cell Migration in Diverse Physical Environments. Proc. Natl. Acad. Sci. USA 2019, 116, 23894–23900. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic Regulation of Cell Growth and Proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436. [Google Scholar] [CrossRef]
- Ortega-Arzola, E.; Higgins, P.M.; Cockell, C.S. The Minimum Energy Required to Build a Cell. Sci. Rep. 2024, 14, 5267. [Google Scholar] [CrossRef] [PubMed]
- Prunier, C.; Chavrier, P.; Boissan, M. Mechanisms of Action of NME Metastasis Suppressors—A Family Affair. Cancer Metastasis Rev. 2023, 42, 1155–1167. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Proust, B.; Horvat, A.; Tadijan, A.; Vlašić, I.; Herak Bosnar, M. Mitochondrial NME6 Influences Basic Cellular Processes in Tumor Cells In Vitro. Int. J. Mol. Sci. 2024, 25, 9580. https://doi.org/10.3390/ijms25179580
Proust B, Horvat A, Tadijan A, Vlašić I, Herak Bosnar M. Mitochondrial NME6 Influences Basic Cellular Processes in Tumor Cells In Vitro. International Journal of Molecular Sciences. 2024; 25(17):9580. https://doi.org/10.3390/ijms25179580
Chicago/Turabian StyleProust, Bastien, Anđela Horvat, Ana Tadijan, Ignacija Vlašić, and Maja Herak Bosnar. 2024. "Mitochondrial NME6 Influences Basic Cellular Processes in Tumor Cells In Vitro" International Journal of Molecular Sciences 25, no. 17: 9580. https://doi.org/10.3390/ijms25179580