
Determining Gene Order Patterns in the Suillus and Boletales through Comparative Analysis of Their Mitogenomes

Abstract
:1. Introduction
2. Results
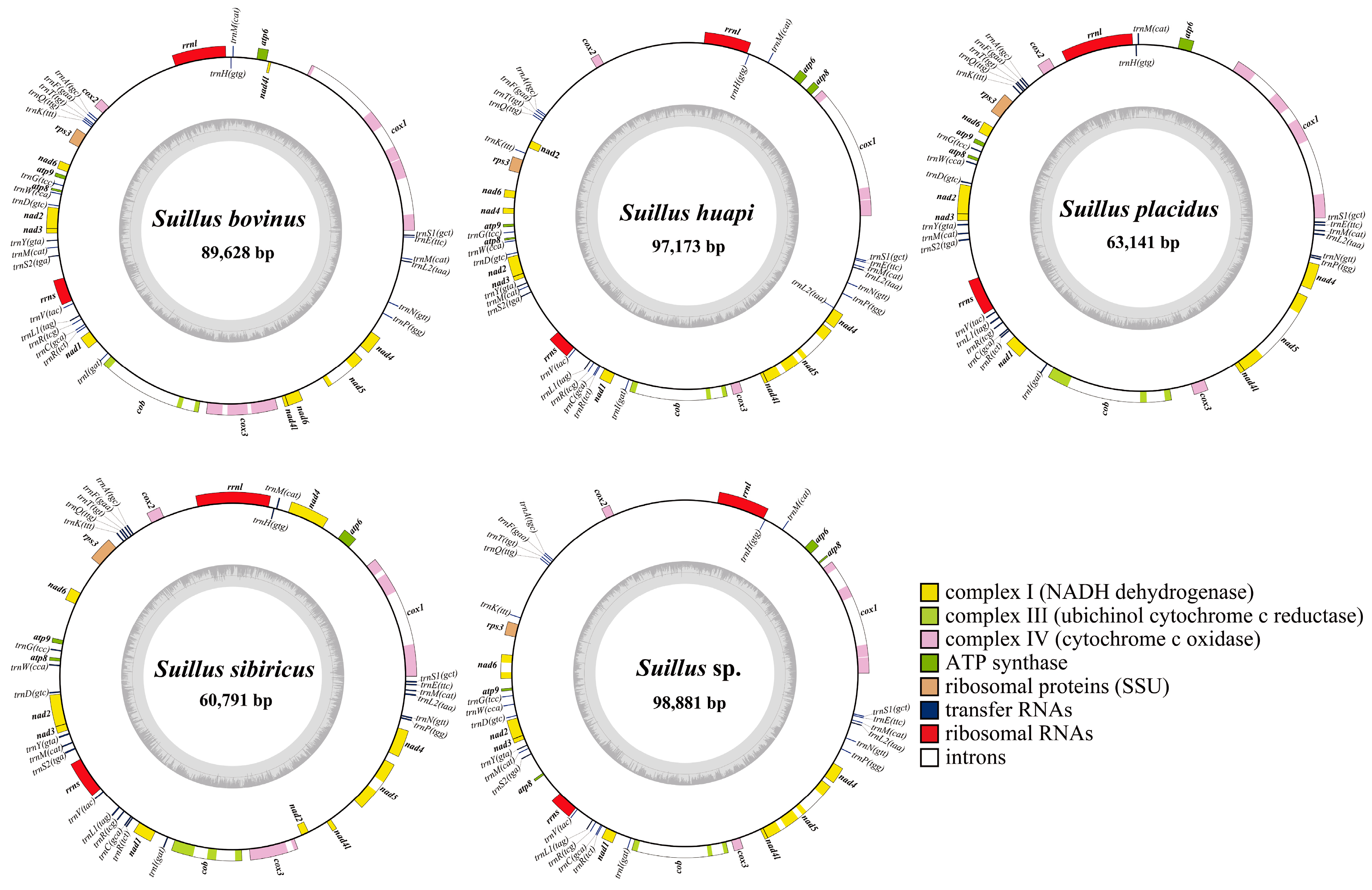
2.1. Features of the Five Suillus Mitogenomes
2.2. PCGs of the Five Suillus Mitogenomes
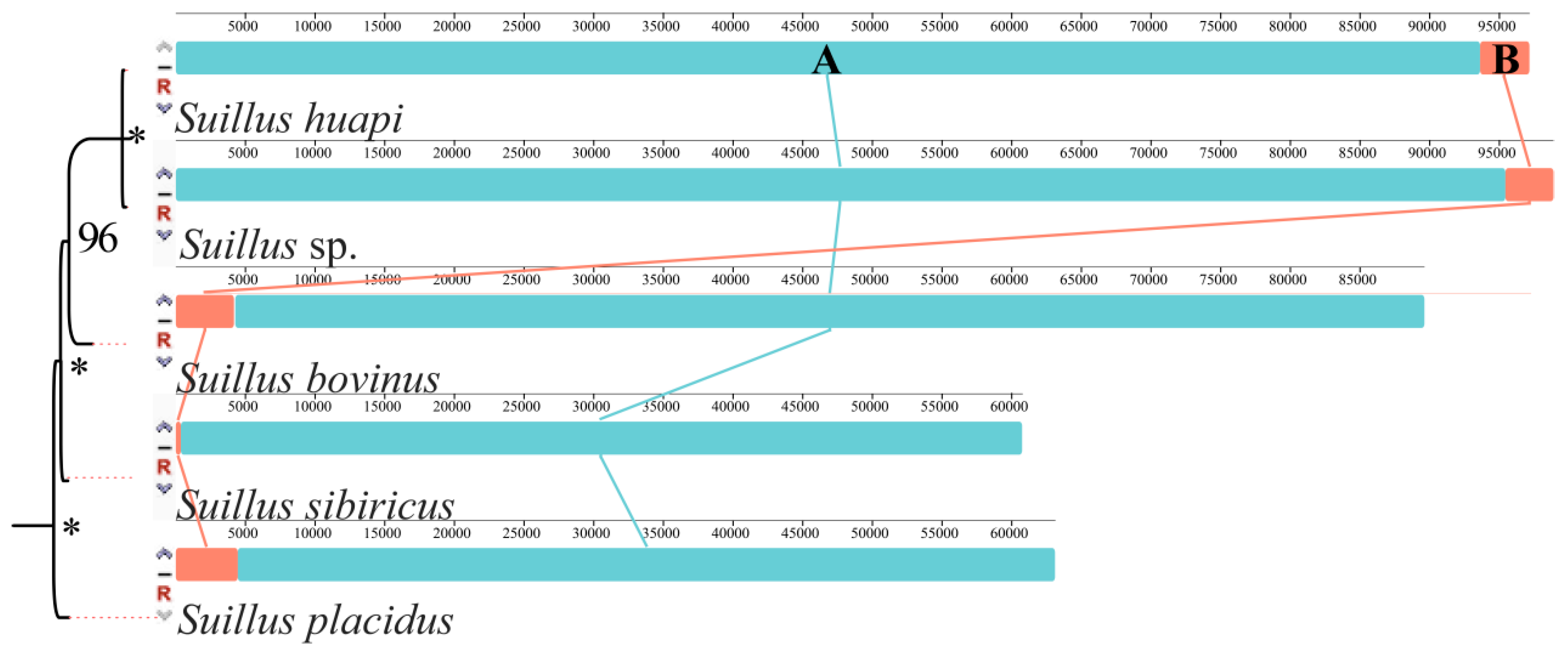
2.3. Gene Rearrangement in the Five Suillus Mitogenomes
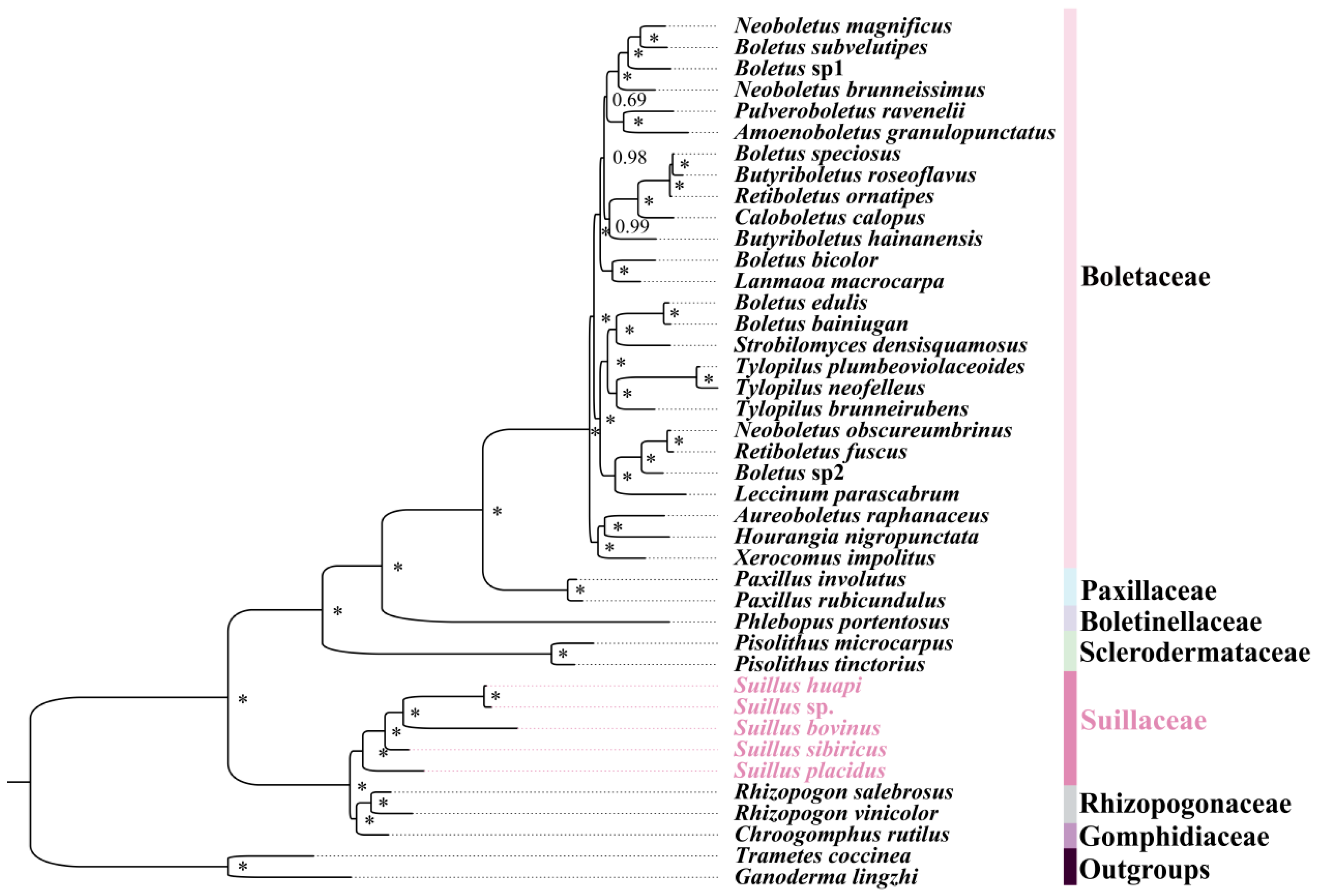
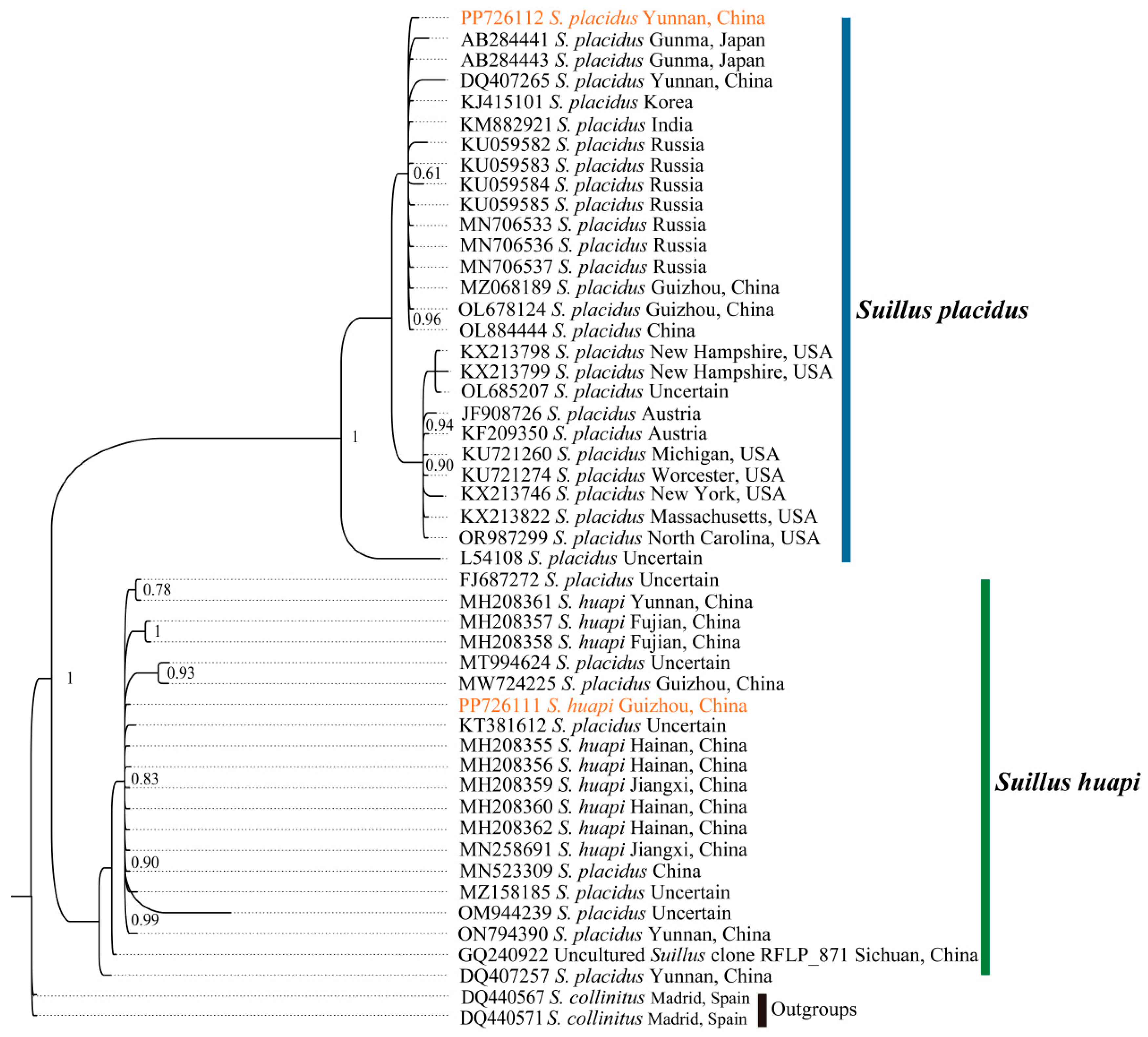
2.4. Phylogenetic Relationships of Boletales
2.5. Gene Order Analysis of Boletales
2.6. Evolutionary Rates of Boletales
3. Discussion
4. Materials and Methods
4.1. Sample Collection, DNA Isolation, and Sequencing
4.2. Assembly, Genome Annotation, and Sequence Analysis
4.3. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Policelli, N.; Bruns, T.D.; Vilgalys, R.; Nuñez, M.A. Suilloid fungi as global drivers of pine invasions. New Phytol. 2019, 222, 714–725. [Google Scholar] [CrossRef]
- Liu, F.Y.; Luo, K.W.; Yu, Z.M.; Co, N.N.; Wu, S.H.; Wu, P.; Fung, K.P.; Kwok, T.T. Suillin from the mushroom Suillus placidus as potent apoptosis inducer in human hepatoma HepG2 cells. Chem. Biol. Interact. 2009, 181, 168–174. [Google Scholar] [CrossRef]
- Dai, Y.C.; Zhou, L.W.; Yang, Z.L.; Wen, H.; Bau, T.; Lit, H. A revised checklist of edible fungi in China. Mycosystema 2010, 29, 1–21. [Google Scholar]
- Klofac, W. A world-wide key to the genus Suillus Weltschlüssel der Gattung Suillus. OestZPilz 2013, 22, 211–278. [Google Scholar]
- Fischer, M.; Jarosch, M.; Binder, M.; Besl, H. Zur Systematik der Boletales: Suillus undverwandte Gattungen. Z. Mykol. 1997, 63, 173–188. [Google Scholar]
- Smith, A.H.; Thiers, H.D. A Contribution toward a Monograph of the North American Species of Suillus (Boletaceae); Lubrecht & Cramer: Ann Arbor, MI, USA, 1964; p. 116. [Google Scholar]
- Gardezi, S.R.A.; Sabir, S.M. Genera Tylopilus, Suillus and Strobilomyces of Azad Jammu and Kashmir, Pakistan. Arch. Phytopathol. Pl. Protect. 2006, 40, 367–375. [Google Scholar] [CrossRef]
- Watling, R. Putative fungus-tree relationships. What do they tell us? Doc. Mycol. 1995, 98–100, 479–486. [Google Scholar]
- Bresinsky, A.; Besl, H. Notizen über Vorkommen und systematische Bewertung von Pigmenten in höheren Pilzen (3)-Untersuchungen an Boletales aus Amerika. Z. Mykol. 1979, 45, 247–264. [Google Scholar]
- Kretzer, A.M.; Bruns, T.D. Use of atp6 in fungal phylogenetics: An example from the Boletales. Molec. Phylogenet. Evol. 1999, 13, 483–492. [Google Scholar] [CrossRef]
- Kretzer, A.M.; Li, Y.; Szaro, T.; Bruns, T.D. Internal transcribed spacer sequences from 38 recognized species of Suillus sensu lato: Phylogenetic and taxonomic implications. Mycologia 1996, 88, 776–785. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Vellinga, E.C.; Bruns, T.D.; Kennedy, P.G. Phylogenetic assessment of global Suillus ITS sequences supports morphologically defined species and reveals synonymous and undescribed taxa. Mycologia 2016, 108, 1216–1228. [Google Scholar] [PubMed]
- Zhang, M.Z.; Xu, J.P.; Callac, P.; Chen, M.Y.; Wu, Q.; Wach, M.; Mata, G.; Zhao, R.L. Insight into the evolutionary and domesticated history of the most widely cultivated mushroom Agaricus bisporus via mitogenome sequences of 361 global strains. BMC Genom. 2023, 24, 182. [Google Scholar] [CrossRef]
- Song, N.; Geng, Y.H.; Li, X.H. The mitochondrial genome of the phytopathogenic fungus Bipolaris sorokiniana and the utility of mitochondrial genome to infer phylogeny of Dothideomycetes. Front. Microbiol. 2020, 11, 863. [Google Scholar] [CrossRef]
- Fan, W.W.; Zhang, S.; Zhang, Y.J. The complete mitochondrial genome of the Chan-hua fungus Isaria cicadae: A tale of intron evolution in Cordycipitaceae. Environ. Microbiol. 2019, 21, 864–879. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.E.; Kwag, Y.N.; Han, S.K.; Lee, D.H.; Kim, C.S. Complete mitochondrial genome sequence of Pulveroboletus ravenelii (Boletales, Basidiomycota). Mitochondrial DNA B Resour. 2022, 7, 1581–1582. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Tu, W.Y.; Bao, Z.J.; Li, L.J.; Li, Q. The first complete mitochondrial genome of edible and medicinal fungus Chroogomphus rutilus (Gomphidiaceae, Boletales) and insights into its phylogeny. Mitochondrial DNA B Resour. 2021, 6, 2355–2357. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Yao, T.; Ren, Y.H.; Ye, J.H.; Qing, Y.; Li, Q.; Gui, M.Y. Evolutionary Insights Into Two Widespread Ectomycorrhizal Fungi (Pisolithus) From Comparative Analysis of Mitochondrial Genomes. Front. Microbiol. 2021, 12, 583129. [Google Scholar] [CrossRef]
- Shi, W.B.; Song, W.C.; Peng, Y.; Wang, S.; Yang, G.W.; Shi, C. The complete mitochondrial genome sequence and annotation of Tylopilus plumbeoviolaceoides T.H. Li, B. Song & Y.H. Shen, 2002 (Boletaceae, Boletoideae). Mitochondrial DNA B Resour. 2022, 7, 999–1000. [Google Scholar]
- Li, Q.; Wu, P.; Li, L.J.; Feng, H.Y.; Tu, W.Y.; Bao, Z.J.; Xiong, C.; Gui, M.Y.; Huang, W.L. The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int. J. Biol. Macromol. 2021, 172, 560–572. [Google Scholar] [CrossRef]
- Li, Q.; Ren, Y.H.; Shi, X.D.; Peng, L.X.; Zhao, J.L.; Song, Y.; Zhao, G. Comparative Mitochondrial Genome Analysis of Two Ectomycorrhizal Fungi (Rhizopogon) Reveals Dynamic Changes of Intron and Phylogenetic Relationships of the Subphylum Agaricomycotina. Int. J. Mol. Sci. 2019, 20, 5167. [Google Scholar] [CrossRef]
- Li, Q.; Ren, Y.H.; Xiang, D.B.; Shi, X.D.; Zhao, J.L.; Peng, L.X.; Zhao, G. Comparative mitogenome analysis of two ectomycorrhizal fungi (Paxillus) reveals gene rearrangement, intron dynamics, and phylogeny of basidiomycetes. IMA Fungus 2020, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Yang, D.; Cao, Y.; Wang, P.; Zhang, Y.; Zhang, K.Q.; Xu, J.; Zhang, Y. The complete mitochondrial genome of the edible Basidiomycete mushroom Phlebopus Portentosus. Mitochondrial DNA Part B 2017, 2, 696–697. [Google Scholar] [CrossRef] [PubMed]
- Formey, D.; Molès, M.; Haouy, A.; Savelli, B.; Bouchez, O.; Bécard, G.; Roux, C. Comparative analysis of mitochondrial genomes of Rhizophagus irregularis–syn. Glomus irregulare–reveals a polymorphism induced by variability generating elements. New Phytol. 2012, 196, 1217–1227. [Google Scholar]
- Himmelstrand, K.; Olson, A.; Brandstrom Durling, M.; Karlsson, M.; Stenlid, J. Intronic and plasmid-derived regions contribute to the large mitochondrial genome sizes of Agaricomycetes. Curr. Genet. 2014, 60, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.M.; Liu, C.C.; Shen, B.M.; Bai, M.; Ling, J.; Chen, G.H.; Mao, Z.C.; Cheng, X.Y.; Xie, B.Y. Analysis of the complete mitochondrial genome of Pochonia chlamydosporia suggests a close relationship to the invertebrate-pathogenic fungi in Hypocreales. BMC Microbiol. 2015, 15, 5. [Google Scholar] [CrossRef]
- Aguileta, G.; De Vienne, D.M.; Ross, O.N.; Hood, M.E.; Giraud, T.; Petit, E. High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 2014, 6, 451–465. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Q.F.; Jin, X.; Chen, Z.Q.; Xiong, C.; Li, P.; Liu, Q.F.; Huang, W.L. Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 2019, 121, 249–260. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, G.Z.; Fang, M.L.; Deng, C.; Zhang, K.Q.; Yu, Z.F.; Xu, J.P. Comparative Analyses of Mitochondrial Genomes Provide Evolutionary Insights Into Nematode-Trapping Fungi. Front. Microbiol. 2020, 11, 617. [Google Scholar] [CrossRef]
- Li, Z.C.; Xie, T.C.; Feng, X.L.; Wang, Z.X.; Lin, C.; Li, G.M.; Li, X.Z.; Qi, J. The First Five Mitochondrial Genomes for the Family Nidulariaceae Reveal Novel Gene Rearrangements, Intron Dynamics, and Phylogeny of Agaricales. Int. J. Mol. Sci. 2023, 24, 12599. [Google Scholar] [CrossRef]
- Xue, R.; Chai, H.; Wang, Y.; Hong, D.; Su, M.S.; Liang, Z.Q.; Zeng, N.K. Species clarification of the locally famous mushroom Suillus placidus from the south of China with description of S. huapi sp. nov. Phytotaxa 2018, 371, 251. [Google Scholar] [CrossRef]
- Galaxy Community. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW), version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xiang, D.B.; Wan, Y.; Wu, Q.; Wu, X.Y.; Ma, C.R.; Song, Y.; Zhao, G.; Huang, W.L. The complete mitochondrial genomes of five important medicinal Ganoderma species: Features, evolution, and phylogeny. Int. J. Biol. Macromol. 2019, 139, 397–408. [Google Scholar] [CrossRef]
- Chen, C.; Li, Q.; Fu, R.T.; Wang, J.; Deng, G.M.; Chen, X.J.; Lu, D.H. Comparative mitochondrial genome analysis reveals intron dynamics and gene rearrangements in two Trametes species. Sci. Rep. 2021, 11, 2569. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef]
- Dereeper, A.; Audic, S.; Claverie, J.M.; Blanc, G. BLAST-Explorer helps you building datasets for phylogenetic analysis. BMC Evol. Biol. 2010, 10, 8. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Jakovli, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Branco, S.; Bi, K.; Liao, H.; Gladieux, P.; Badouin, H.; Ellison, C.E.; Nguyen, N.H.; Vilgalys, R.; Peay, K.G.; Taylor, J.W.; et al. Continental-level population differentiation and environmental adaptation in the mushroom Suillus brevipes. Mol. Ecol. 2017, 26, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Mueller, G.M.; Shi, X.; Liu, P. Two new species in the Suillus spraguei complex from China. Mycologia 2017, 109, 296–307. [Google Scholar] [CrossRef]
- Bourke, B.P.; Justi, S.A.; Caicedo-Quiroga, L.; Pecor, D.B.; Wilkerson, R.C.; Linton, Y.M. Phylogenetic analysis of the Neotropical Albitarsis Complex based on mitogenome data. Parasites Vectors 2021, 14, 589. [Google Scholar] [CrossRef]
- Françoso, E.; Zuntini, A.R.; Ricardo, P.C.; Santos, P.K.F.; De Souza Araujo, N.; Silva, J.P.N.; Gonçalves, L.T.; Brito, R.; Gloag, R.; Taylor, B.A.; et al. Rapid evolution, rearrangements and whole mitogenome duplication in the Australian stingless bees Tetragonula (Hymenoptera: Apidae): A steppingstone towards understanding mitochondrial function and evolution. Int. J. Biol. Macromol. 2023, 242, 124568. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Length (bp) | GC Rate (%) | AT Skew | GC Skew | tRNAs | Introns |
|---|---|---|---|---|---|---|
| S. bovinus | 89,628 | 20.7 | −0.009 | 0.060 | 25 | 9 |
| S. huapi | 97,173 | 19.7 | 0.006 | 0.073 | 26 | 7 |
| S. placidus | 63,141 | 22.5 | −0.010 | 0.067 | 25 | 6 |
| S. sibiricus | 60,791 | 21.3 | −0.024 | 0.066 | 25 | 6 |
| Suillus sp. | 98,881 | 19.6 | 0.002 | 0.073 | 25 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, J.; Wang, X.; Long, Y.; Gao, Z.; Zhang, G.; Guo, Z.; Wang, G.; Xu, G.; Wang, Y.; Liu, H. Determining Gene Order Patterns in the Suillus and Boletales through Comparative Analysis of Their Mitogenomes. Int. J. Mol. Sci. 2024, 25, 9597. https://doi.org/10.3390/ijms25179597
Tao J, Wang X, Long Y, Gao Z, Zhang G, Guo Z, Wang G, Xu G, Wang Y, Liu H. Determining Gene Order Patterns in the Suillus and Boletales through Comparative Analysis of Their Mitogenomes. International Journal of Molecular Sciences. 2024; 25(17):9597. https://doi.org/10.3390/ijms25179597
Chicago/Turabian StyleTao, Jiawei, Xianyi Wang, Yaohang Long, Zexin Gao, Gongyou Zhang, Zhongyao Guo, Guoyu Wang, Guangyin Xu, Yaping Wang, and Hongmei Liu. 2024. "Determining Gene Order Patterns in the Suillus and Boletales through Comparative Analysis of Their Mitogenomes" International Journal of Molecular Sciences 25, no. 17: 9597. https://doi.org/10.3390/ijms25179597