
Inflammatory Intracellular Signaling in Neurons Is Influenced by Glial Soluble Factors in iPSC-Based Cell Model of PARK2-Associated Parkinson’s Disease

 , , ,
, , ,  ,
,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
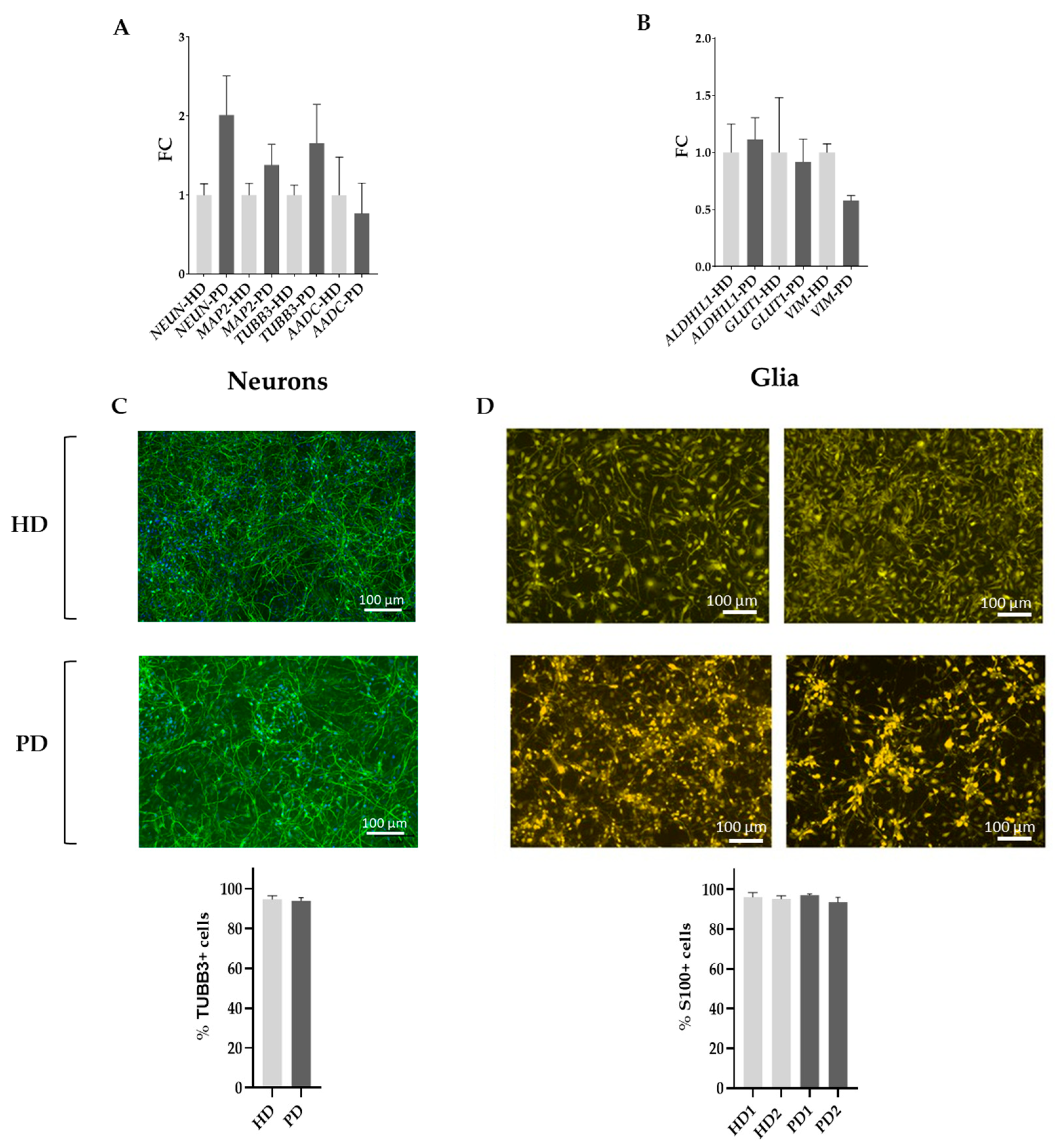
2.1. Characterization of the Initial Cell Composition and Differentiation Status of the Cell Cultures Used in This Study
2.1.1. Neuronal Cell Cultures
2.1.2. Glial Cell Cultures
- ○
- HD0—HD neurons in the base neuronal medium.
- ○
- PD0—PD neurons in the base neuronal medium.
- ○
- HD1—HD neurons in the base glial medium.
- ○
- PD1—PD neurons in the base glial medium.
- ○
- HD2—HD neurons in the HD glial mixed medium.
- ○
- PD2—PD neurons in the HD glial mixed medium.
- ○
- HD3—HD neurons in the PD glial mixed medium.
- ○
- PD3—PD neurons in the PD glial mixed medium.
2.2. Full-Transcriptome Profiling Using High-Throughput RNA Sequencing (RNAseq) and Bioinformatics Data Analysis
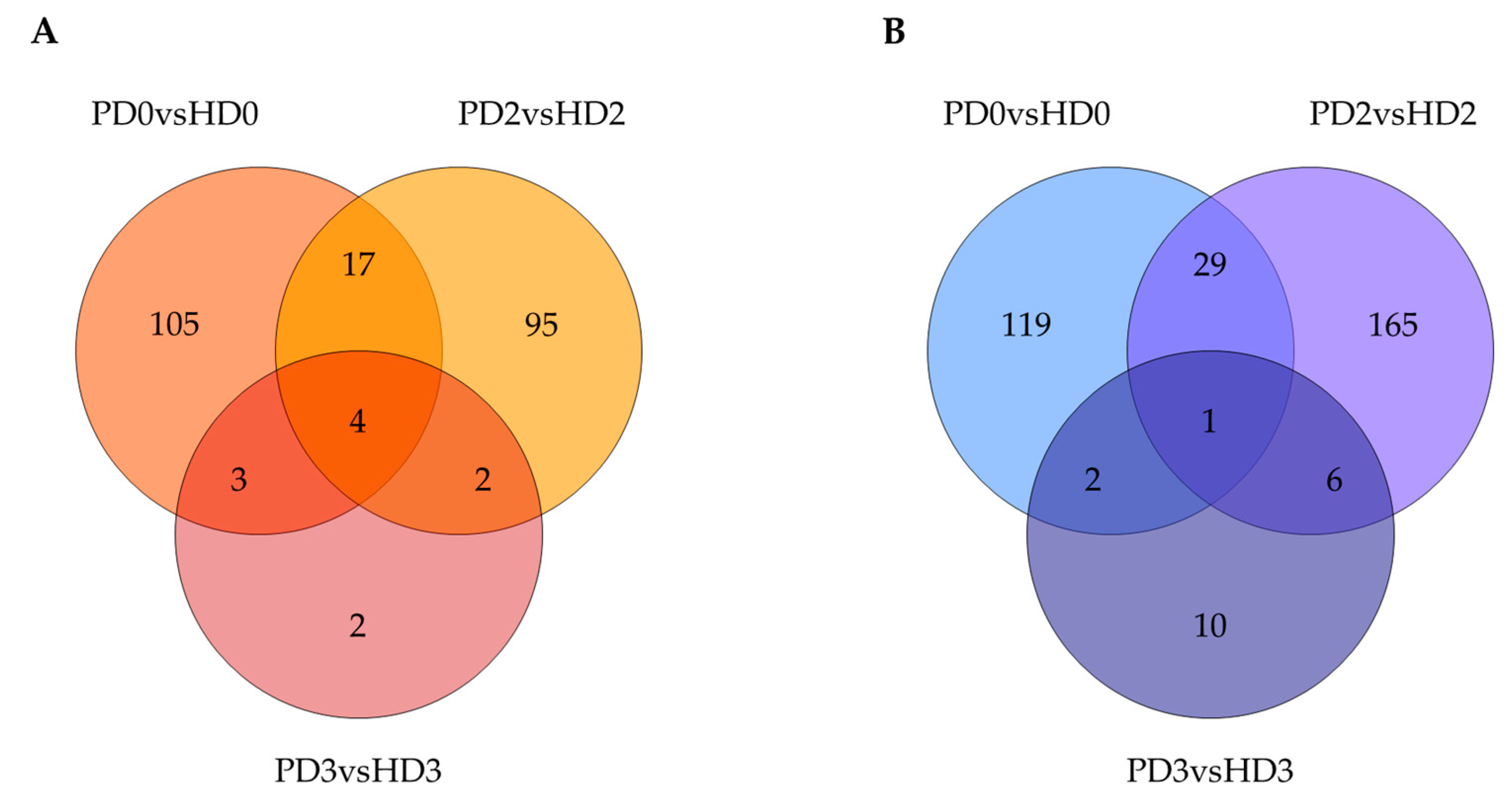
2.3. Identification of DEGs Related to the Disease Status of Neurons or to the Glial Medium Type
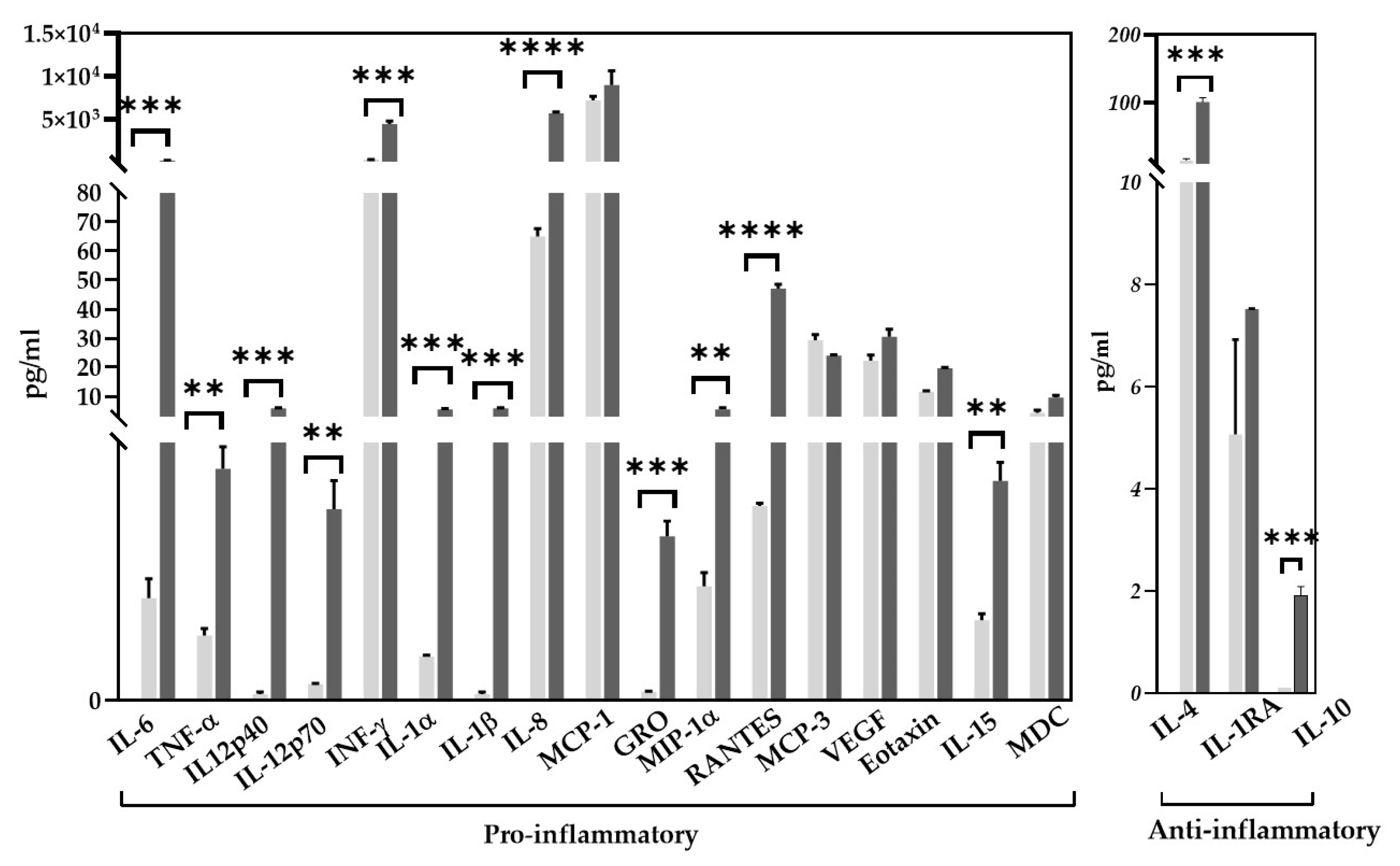
2.4. Determination of Cytokine/Chemokine Composition of Glial Mixed Supernatants
2.5. Determination of the Neuronal Expression of Genes Encoding Receptors to Corresponding Glial-Derived Cytokines Found in HD and PD Glial Mixed Supernatants
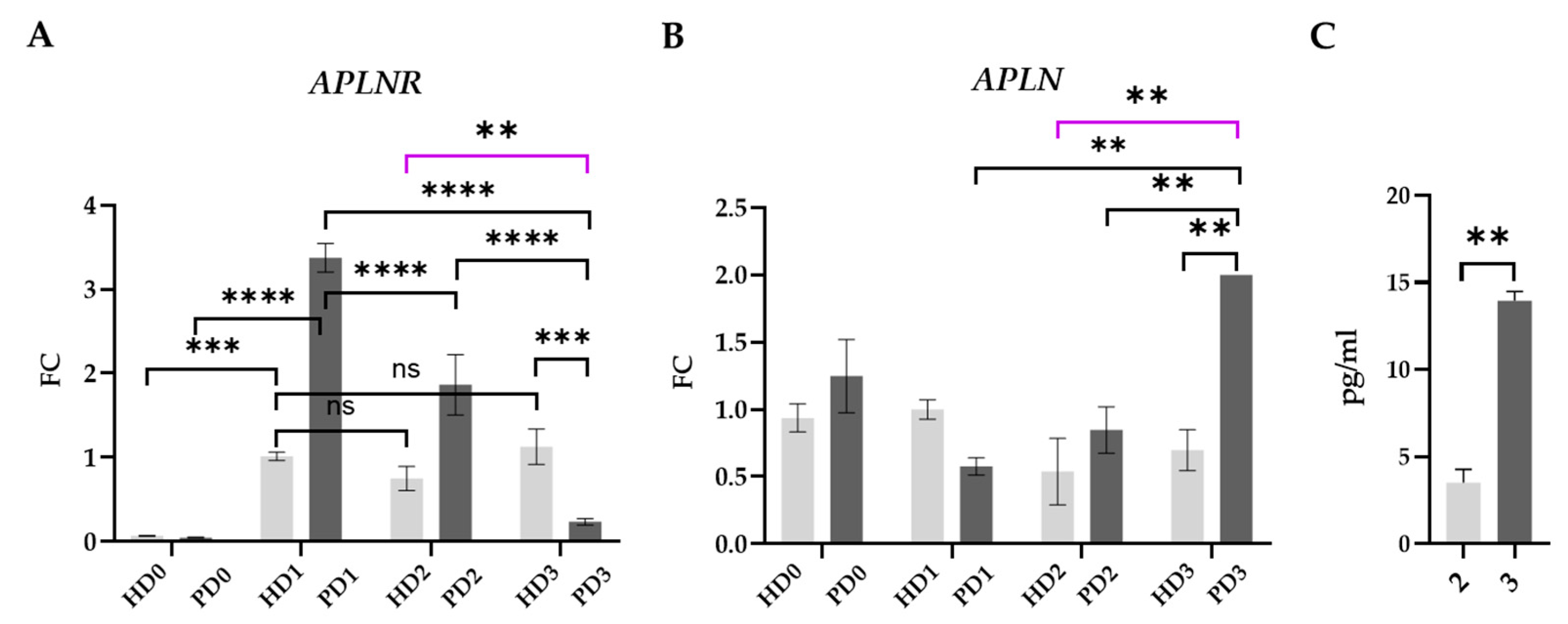
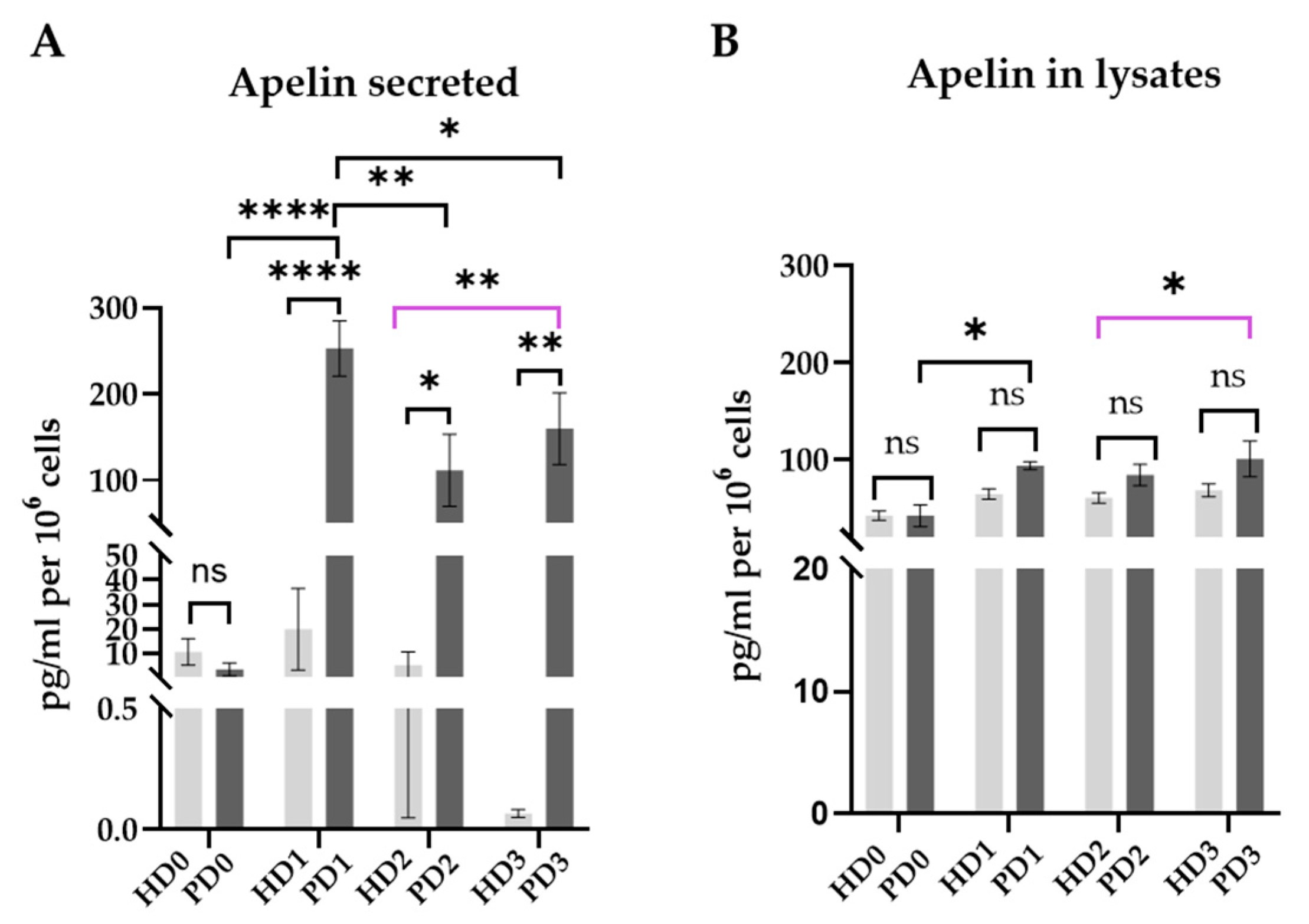
2.6. Apelin/APLNR System
- (i)
- PD glial mixed media contained comparatively higher concentrations of a number of pro-inflammatory and anti-inflammatory cytokines and chemokines than HD glial mixed medium.
- (ii)
- The most genes encoding receptors to the studied cytokines were expressed on the same level in PD and HD neurons, and that was not affected by soluble factors of glial mixed media. Thus, no signs of feedback regulation were detected.
- (iii)
- The inflammatory pathways enriched in PD2 vs. HD2, in PD3 vs. HD2, and in HD3 vs. HD2 demonstrated differences dependent both on the neurons’ genetic status and on the glial soluble factors. Among the main enriched pathways, MAPK signaling and those related to cytokines (cytokine–receptor interactions, responses to cytokine, and cytokine production) were detected.
- (iv)
- The ‘HD neuron/HD glia’ system might be characterized as more reactive than the PD one, as HD neurons and soluble factors of HD glia demonstrated greater ability to interact with each other.
- (v)
- If one or both of the components, neurons or glia, is Parkin-deficient, humoral interactions between the cell types led to a general suppression of the inflammatory gene network in neurons compared to the HD system.
- (vi)
- APLNR expression was up-regulated in HD and PD neurons as a stress response. In cases of PD neurons, this up-regulation was stronger and was accompanied by increased synthesis and release of apelin into intracellular fluid. In PD neurons, but not in HD cells, APLNR expression and apelin secretion were down-regulated by glial soluble factors, both HD and PD. PD glial secretome to the greatest extent depressed APLNR in PD neurons, which was accompanied by a high level of APLN transcription and apelin release.
3. Materials and Methods
3.1. iPSC Differentiation in Neuronal and Glial Direction and Cultivation of Neurons
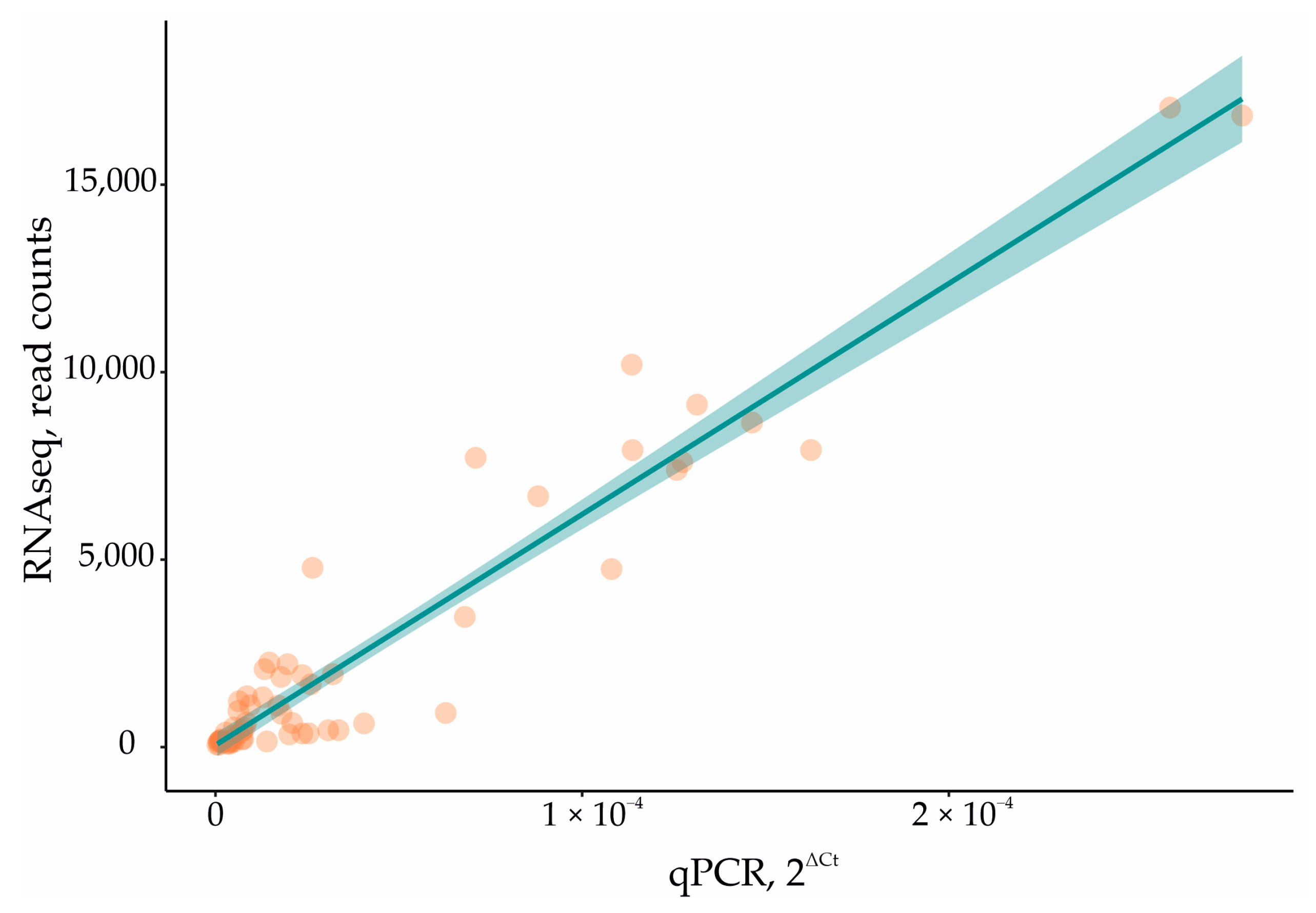
3.2. Full-Transcriptome Profiling Using High-Throughput RNA Sequencing (RNAseq) Technology and Bioinformatics Data Analysis
3.3. Immunocytochemistry
3.4. Multiplex Analysis of Proteins
3.5. ELISA
3.6. Quantitative Real-Time PCR (qPCR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Abbreviations
| PD | Parkinson’s disease |
| HD | healthy donor |
| iPSC | induced pluripotent stem cell |
| DA neurons | dopaminergic neurons |
| CNS | central nervous system |
| DEG | differentially expressed gene |
| FC | fold change |
References
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Bisaglia, M. Oxidative Stress and Neuroinflammation in Parkinson’s Disease: The Role of Dopamine Oxidation Products. Antioxidants 2023, 12, 955. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Bougea, A.; Piperi, C. Impact of the Apelin/APJ Axis in the Pathogenesis of Parkinson’s Disease with Therapeutic Potential. J. Neurosci. Res. 2021, 99, 2117–2133. [Google Scholar] [CrossRef] [PubMed]
- Puspita, L.; Chung, S.Y.; Shim, J. Oxidative Stress and Cellular Pathologies in Parkinson’s Disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, R.; Nisa Awan, M.U.; Bai, J. The Mechanism and Function of Glia in Parkinson’s Disease. Front. Cell. Neurosci. 2022, 16, 903469. [Google Scholar] [CrossRef]
- Chiareli, R.A.; Carvalho, G.A.; Marques, B.L.; Mota, L.S.; Oliveira-Lima, O.C.; Gomes, R.M.; Birbrair, A.; Gomez, R.S.; Simão, F.; Klempin, F.; et al. The Role of Astrocytes in the Neurorepair Process. Front. Cell Dev. Biol. 2021, 9, 665795. [Google Scholar] [CrossRef]
- Alqawlaq, S.; Livne-Bar, I.; Chan, D.; Sivak, J.M. What Happens When Astrocytes Talk to Neurons? A Proteomic PI3K Interactome Study. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1486. [Google Scholar]
- Spocter, M.A.; Hopkins, W.D.; Barks, S.K.; Bianchi, S.; Hehmeyer, A.E.; Anderson, S.M.; Stimpson, C.D.; Fobbs, A.J.; Hof, P.R.; Sherwood, C.C. Neuropil Distribution in the Cerebral Cortex Differs between Humans and Chimpanzees. J. Comp. Neurol. 2012, 520, 2917–2929. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.; Zorec, R. Astrocytes as Secretory Cells of the Central Nervous System: Idiosyncrasies of Vesicular Secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef]
- Jha, M.K.; Kim, J.-H.; Song, G.J.; Lee, W.-H.; Lee, I.-K.; Lee, H.-W.; An, S.S.A.; Kim, S.; Suk, K. Functional Dissection of Astrocyte-Secreted Proteins: Implications in Brain Health and Diseases. Prog. Neurobiol. 2018, 162, 37–69. [Google Scholar] [CrossRef]
- Chang, M.-Y.; Son, H.; Lee, Y.-S.; Lee, S.-H. Neurons and Astrocytes Secrete Factors That Cause Stem Cells to Differentiate into Neurons and Astrocytes, Respectively. Mol. Cell. Neurosci. 2003, 23, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, S.F.; Bortolotto, V.; Canonico, P.L.; Sortino, M.A.; Grilli, M. Astrocyte-Derived Paracrine Signals: Relevance for Neurogenic Niche Regulation and Blood–Brain Barrier Integrity. Front. Pharmacol. 2019, 10, 1346. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.; Kim, S.U. Human Astrocytes: Secretome Profiles of Cytokines and Chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef]
- Dzamko, N. Cytokine Activity in Parkinson’s Disease. Neuronal Signal. 2023, 7, NS20220063. [Google Scholar] [CrossRef]
- Klegeris, A. Targeting Neuroprotective Functions of Astrocytes in Neuroimmune Diseases. Expert Opin. Ther. Targets 2021, 25, 237–241. [Google Scholar] [CrossRef]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin Mitochondrial Quality Control: A Source of Regional Vulnerability in Parkinson’s Disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef]
- Ben Haim, L.; Carrillo-de Sauvage, M.-A.; Ceyzériat, K.; Escartin, C. Elusive Roles for Reactive Astrocytes in Neurodegenerative Diseases. Front. Cell. Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Petsko, G.A.; Studer, L. Induced Pluripotent Stem Cells: A Tool for Modeling Parkinson’s Disease. Trends Neurosci. 2022, 45, 608–620. [Google Scholar] [CrossRef]
- Whiteley, J.T.; Fernandes, S.; Sharma, A.; Mendes, A.P.D.; Racha, V.; Benassi, S.K.; Marchetto, M.C. Reaching into the Toolbox: Stem Cell Models to Study Neuropsychiatric Disorders. Stem Cell Rep. 2022, 17, 187–210. [Google Scholar] [CrossRef]
- Novosadova, E.V.; Nekrasov, E.D.; Chestkov, I.V.; Surdina, A.V.; Vasina, E.M.; Bogomazova, A.N.; Manuilova, E.S.; Arsenyeva, E.L.; Simonova, V.V.; Konovalova, E.V.; et al. A Platform for Studying Molecular and Cellular Mechanisms of Parkinson’s Disease Based on Human Induced Pluripotent Stem Cells. Sovrem. Tehnol. Med. 2016, 8, 157–166. [Google Scholar] [CrossRef]
- Novosadova, E.; Anufrieva, K.; Kazantseva, E.; Arsenyeva, E.; Fedoseyeva, V.; Stepanenko, E.; Poberezhniy, D.; Illarioshkin, S.; Novosadova, L.; Gerasimova, T.; et al. Transcriptome datasets of neural progenitors and neurons differentiated from induced pluripotent stem cells of healthy donors and Parkinson’s disease patients with mutations in the PARK2 gene. Data Brief 2022, 41, 107958. [Google Scholar] [CrossRef] [PubMed]
- Shuvalova, L.D.; Eremeev, A.V.; Bogomazova, A.N.; Novosadova, E.V.; Zerkalenkova, E.A.; Olshanskaya, Y.V.; Fedotova, E.Y.; Glagoleva, E.S.; Illarioshkin, S.N.; Lebedeva, O.S.; et al. Generation of Induced Pluripotent Stem Cell Line RCPCMi004-A Derived from Patient with Parkinson’s Disease with Deletion of the Exon 2 in PARK2 Gene. Stem Cell Res. 2020, 44, 101733. [Google Scholar] [CrossRef]
- Gerasimova, T.; Stepanenko, E.; Novosadova, L.; Arsenyeva, E.; Shimchenko, D.; Tarantul, V.; Grivennikov, I.; Nenasheva, V.; Novosadova, E. Glial Cultures Differentiated from IPSCs of Patients with PARK2-Associated Parkinson’s Disease Demonstrate a Pro-Inflammatory Shift and Reduced Response to TNFα Stimulation. Int. J. Mol. Sci. 2023, 24, 2000. [Google Scholar] [CrossRef] [PubMed]
- Novosadova, E.V.; Arsenyva, E.L.; Antonov, S.A.; Kazantseva, E.A.; Novosadova, L.V.; Kurko, O.D.; Illarioshkin, S.N.; Tarantul, V.Z.; Grivennikov, I.A. Generation and Characteristics of Glial Cells from Induced Human Pluripotent Stem Cells. Neurochemistry 2020, 37, 358–367. [Google Scholar] [CrossRef]
- Solomon, B.; Gavriel, Y.; Rabinovich-Nikitin, I. Inhibition of CXCR4/CXCL12 Signaling: A Translational Perspective for Alzheimer’s Disease Treatment. Neural Regener. Res. 2022, 17, 108. [Google Scholar] [CrossRef]
- Shimoji, M.; Pagan, F.; Healton, E.B.; Mocchetti, I. CXCR4 and CXCL12 Expression Is Increased in the Nigro-Striatal System of Parkinson’s Disease. Neurotox. Res. 2009, 16, 318–328. [Google Scholar] [CrossRef]
- Aimé, P.; Sun, X.; Zareen, N.; Rao, A.; Berman, Z.; Volpicelli-Daley, L.; Bernd, P.; Crary, J.F.; Levy, O.A.; Greene, L.A. Trib3 Is Elevated in Parkinson’s Disease and Mediates Death in Parkinson’s Disease Models. J. Neurosci. 2015, 35, 10731–10749. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Kam, T.-I.; Wang, H.; Neifert, S.; Chou, S.-C.; Kumar, M.; Brahmachari, S.; Jhaldiyal, A.; Hinkle, J.T.; Akkentli, F.; et al. Neuronal NLRP3 Is a Parkin Substrate That Drives Neurodegeneration in Parkinson’s Disease. Neuron 2022, 110, 2422–2437. [Google Scholar] [CrossRef]
- Ayimugu, A.; Sarihan, M.; Kasap, M.; Akpinar, G. Investigation of the Involvement of Parkin in Parkinson’s Disease and Cancer by Monitoring the Changes in SH-SY5Y Cells at the Nuclear Proteome Level. Anticancer Res. 2020, 40, 3169–3190. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A Master Regulator of Cellular Responses in Inflammation, Injury Resolution, and Tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Zhang, F.; Pan, L.; Lian, C.; Xu, Z.; Chen, H.; Lai, W.; Liang, X.; Liu, Q.; Wu, H.; Wang, Y.; et al. ICAM-1 May Promote the Loss of Dopaminergic Neurons by Regulating Inflammation in MPTP-induced Parkinson’s Disease Mouse Models. Brain Res. Bull. 2024, 214, 110989. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.-E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef]
- Ma, C.; Wei, X.; Wang, F.; Zhang, T.; Jiang, Y.; Meng, Z.; Zhang, Z. Tumor Necrosis Factor α-Induced Protein 3 Mediates Inflammation and Neuronal Autophagy in Parkinson’s Disease via the NFκB and MTOR Pathways. Neurosci. Lett. 2023, 805, 137223. [Google Scholar] [CrossRef] [PubMed]
- Perga, S.; Martire, S.; Montarolo, F.; Navone, N.D.; Calvo, A.; Fuda, G.; Marchet, A.; Leotta, D.; Chiò, A.; Bertolotto, A. A20 in Multiple Sclerosis and Parkinson’s Disease: Clue to a Common Dysregulation of Anti-Inflammatory Pathways? Neurotox. Res. 2017, 32, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Carow, B.; Rottenberg, M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Sood, V.; Lata, S.; Ramachandran, V.G.; Banerjea, A.C. Suppressor of Cytokine Signaling 3 (SOCS3) Degrades P65 and Regulate HIV-1 Replication. Front. Microbiol. 2019, 10, 114. [Google Scholar] [CrossRef]
- Tiwari, P.C.; Pal, R. The Potential Role of Neuroinflammation and Transcription Factors in Parkinson Disease. Psychoneuroimmunology 2017, 19, 71–80. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Ammit, A.J. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Anti-Inflammatory Responses. Front. Immunol. 2019, 10, 1446. [Google Scholar] [CrossRef]
- Bhore, N.; Wang, B.J.; Chen, Y.W.; Liao, Y.F. Critical Roles of Dual-Specificity Phosphatases in Neuronal Proteostasis and Neurological Diseases. Int. J. Mol. Sci. 2017, 18, 1963. [Google Scholar] [CrossRef]
- Subbannayya, Y.; Pinto, S.M.; Bösl, K.; Prasad, T.S.K.; Kandasamy, R.K. Dynamics of Dual Specificity Phosphatases and Their Interplay with Protein Kinases in Immune Signaling. Int. J. Mol. Sci. 2019, 20, 2086. [Google Scholar] [CrossRef]
- Zenz, R.; Eferl, R.; Scheinecker, C.; Redlich, K.; Smolen, J.; Schonthaler, H.B.; Kenner, L.; Tschachler, E.; Wagner, E.F. Activator Protein 1 (Fos/Jun) Functions in Inflammatory Bone and Skin Disease. Arthritis Res. Ther. 2007, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.; Raivich, G.; Anderson, P.N. Activating Transcription Factor 3 and the Nervous System. Front. Mol. Neurosci. 2012, 5, 7. [Google Scholar] [CrossRef]
- Liu, H.; Zeng, L.; Yang, Y.; Guo, C.; Wang, H. Bcl-3: A Double-Edged Sword in Immune Cells and Inflammation. Front. Immunol. 2022, 13, 847699. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, K.; Lee, H.; Jeon, J.; Lee, J.; Yoon, J. The Protein-Protein Interaction Network of Hereditary Parkinsonism Genes Is a Hierarchical Scale-Free Network. Yonsei Med. J. 2022, 63, 724. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.L.; Hilal, S.; Chong, J.P.C.; Ng, Y.X.; Liew, O.W.; Xu, X.; Ikram, M.K.; Venketasubramanian, N.; Richards, A.M.; Lai, M.K.P.; et al. Growth Differentiation Factor-15 and White Matter Hyperintensities in Cognitive Impairment and Dementia. Medicine 2016, 95, e4566. [Google Scholar] [CrossRef]
- Machado, V.; Haas, S.J.; von Bohlen Und Halbach, O.; Wree, A.; Krieglstein, K.; Unsicker, K.; Spittau, B. Growth/differentiation Factor-15 Deficiency Compromises Dopaminergic Neuron Survival and Microglial Response in the 6-hydroxydopamine Mouse Model of Parkinson’s Disease. Neurobiol. Dis. 2016, 88, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Griñán-Ferré, C.; Jarne-Ferrer, J.; Bellver-Sanchis, A.; Ribalta-Vilella, M.; Barroso, E.; Salvador, J.M.; Jurado-Aguilar, J.; Palomer, X.; Vázquez-Carrera, M.; Pallàs, M. Deletion of Gadd45a Expression in Mice Leads to Cognitive and Synaptic Impairment Associated with Alzheimer’s Disease Hallmarks. Int. J. Mol. Sci. 2024, 25, 2595. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Collier, T.J.; Cole-Strauss, A.; Grabinski, T.; Mattingly, Z.R.; Winn, M.E.; Steece-Collier, K.; Sortwell, C.E.; Manfredsson, F.P.; Lipton, J.W. The Longitudinal Transcriptomic Response of the Substantia Nigra to Intrastriatal 6-hydroxydopamine Reveals Significant Upregulation of Regeneration-associated Genes. PLoS ONE 2015, 10, 0127768. [Google Scholar] [CrossRef]
- Unschuld, P.G.; Dächsel, J.; Darios, F.; Kohlmann, A.; Casademunt, E.; Lehmann-Horn, K.; Dichgans, M.; Ruberg, M.; Brice, A.; Gasser, T.; et al. Parkin Modulates Gene Expression in Control and Ceramide-Treated PC12 Cells. Mol. Biol. Rep. 2006, 33, 13–32. [Google Scholar] [CrossRef]
- Wei, C.; Li, Y.; Zhu, Z.; Jia, D.; Fan, M.; Li, T.; Wang, X.; Li, Z.; Ma, H. Inhibition of Activator Protein 1 Attenuates Neuroinflammation and Brain Injury after Experimental Intracerebral Hemorrhage. CNS Neurosci. Ther. 2019, 25, 1182–1188. [Google Scholar] [CrossRef]
- Wang, H.; Chen, G.; Ahn, E.H.; Xia, Y.; Kang, S.S.; Liu, X.; Liu, C.; Han, M.H.; Chen, S.; Ye, K. C/EBPβ/AEP Is Age-Dependently Activated in Parkinson’s Disease and Mediates α-Synuclein in the Gut and Brain. ProQuest 2023, 9, 1. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-κB-Mediated Neuroinflammation in Parkinson’s Disease and Potential Therapeutic Effect of Polyphenols. Neurotox. Res. 2020, 37, 491–507. [Google Scholar] [CrossRef]
- Perner, C.; Perner, F.; Gaur, N.; Zimmermann, S.; Witte, O.W.; Heidel, F.H.; Grosskreutz, J.; Prell, T. Plasma VCAM1 Levels Correlate with Disease Severity in Parkinson’s Disease. J. Neuroinflamm. 2019, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Letsiou, E.; Sammani, S.; Wang, H.; Belvitch, P.; Dudek, S.M. Parkin regulates lipopolysaccharide-induced proinflammatory responses in acute lung injury. Transl. Res. J. Lab. Clin. Med. 2017, 181, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Xu, Y.; Liu, Z.; Wang, Y.; Xu, X.; Li, C.; Li, S.; Zhang, J.; Xiong, T.; Cao, W.; et al. IGF2 Inhibits Hippocampal Over-Activated Microglia and Alleviates Depression-like Behavior in LPS- Treated Male Mice. Brain Res. Bull. 2023, 194, 1–12. [Google Scholar] [CrossRef]
- Sepúlveda, D.; Grunenwald, F.; Vidal, A.; Troncoso-Escudero, P.; Cisternas-Olmedo, M.; Villagra, R.; Vergara, P.; Aguilera, C.; Nassif, M.; Vidal, R.L. Insulin-like Growth Factor 2 and Autophagy Gene Expression Alteration Arise as Potential Biomarkers in Parkinson’s Disease. Sci. Rep. 2022, 12, 2038. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. TGF-β as a Key Modulator of Astrocyte Reactivity: Disease Relevance and Therapeutic Implications. Biomedicines 2022, 10, 1206. [Google Scholar] [CrossRef]
- Meyers, E.A.; Kessler, J.A. TGF-β Family Signaling in Neural and Neuronal Differentiation, Development, and Function. Cold Spring Harb. Perspect. Biol. 2017, 9, a022244. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Pattison, M.B.; Midkiff, C.C. The FGF/FGFR System in the Microglial Neuroinflammation with Borrelia Burgdorferi: Likely Intersectionality with Other Neurological Conditions. J. Neuroinflamm. 2023, 20, 10. [Google Scholar] [CrossRef]
- Tsuchida, T.; Mano, T.; Koshi-Mano, K.; Bannai, T.; Matsubara, T.; Yamashita, S.; Ushijima, T.; Nagata, K.; Murayama, S.; Toda, T.; et al. Methylation Changes and Aberrant Expression of FGFR3 in Lewy Body Disease Neurons. Brain Res. 2018, 1697, 59–66. [Google Scholar] [CrossRef]
- Enogieru, A.B.; Omoruyi, S.I.; Hiss, D.C.; Ekpo, O.E. GRP78/BIP/HSPA5 as a Therapeutic Target in Models of Parkinson’s Disease: A Mini Review. Adv. Pharmacol. Sci. 2019, 2019, 2706783. [Google Scholar] [CrossRef]
- Choi, D.-J.; An, J.; Jou, I.; Park, S.M.; Joe, E.-H. A Parkinson’s Disease Gene, DJ-1, Regulates Anti-Inflammatory Roles of Astrocytes through Prostaglandin D2 Synthase Expression. Neurobiol. Dis. 2019, 127, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.I.; Vlasov, I.N.; Partevian, S.A.; Rosinskaya, A.V.; Rybolovlev, I.N.; Slominsky, P.A.; Shadrina, M.I.; Alieva, A.K. Transcriptome Profiling Reveals Differential Expression of Circadian Behavior Genes in Peripheral Blood of Monozygotic Twins Discordant for Parkinson’s Disease. Cells 2022, 11, 2599. [Google Scholar] [CrossRef]
- Danelon, V.; Garret-Thomson, S.C.; Almo, S.C.; Lee, F.S.; Hempstead, B.L. Immune Activation of the P75 Neurotrophin Receptor: Implications in Neuroinflammation. Front. Mol. Neurosci. 2023, 16, 1305574. [Google Scholar] [CrossRef]
- Ali, N.H.; Al-kuraishy, H.M.; Al-Gareeb, A.I.; Alnaaim, S.A.; Saad, H.M.; Batiha, G.E. The Molecular Pathway of P75 Neurotrophin Receptor (P75NTR) in Parkinson’s Disease: The Way of New Inroads. Mol. Neurobiol. 2023, 61, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, L.; Xie, J.; Shi, L. The Emerging Role of Neuropeptides in Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 646–726. [Google Scholar] [CrossRef] [PubMed]
- Yildirim-Balatan, C.; Fenyi, A.; Besnault, P.; Gomez, L.; Sepulveda-Diaz, J.E.; Michel, P.P.; Melki, R.; Hunot, S. Parkinson’s Disease-derived α-synuclein Assemblies Combined with Chronic-type Inflammatory Cues Promote a Neurotoxic Microglial Phenotype. J. Neuroinflamm. 2024, 21, 54. [Google Scholar] [CrossRef]
- Nam, M.-H.; Sa, M.; Ju, Y.H.; Park, M.G.; Lee, C.J. Revisiting the Role of Astrocytic MAOB in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 4453. [Google Scholar] [CrossRef]
- Jiang, H.; Jiang, Q.; Liu, W.; Feng, J. Parkin suppresses the expression of monoamine oxidases. J. Biol. Chem. 2006, 281, 8591–8599. [Google Scholar] [CrossRef]
- Michelis, R.; Milhem, L.; Galouk, E.; Stemer, G.; Aviv, A.; Tadmor, T.; Shehadeh, M.; Shvidel, L.; Barhoum, M.; Braester, A. Increased Serum Level of Alpha-2 Macroglobulin and Its Production by B-Lymphocytes in Chronic Lymphocytic Leukemia. Front. Immunol. 2022, 13, 953644. [Google Scholar] [CrossRef]
- Aisha, Z.; Lei, J.; Zhang, Y.; Ma, J. EEF1A1 Is Involved the Regulating Neuroinflammatory Processes in Parkinson’s Disease. J. Integr. Neurosci. 2023, 22, 122. [Google Scholar] [CrossRef] [PubMed]
- Thichanpiang, P.; Harper, S.J.; Wongprasert, K.; Bates, D.O. TNF-α-induced ICAM-1 Expression and Monocyte Adhesion in Human RPE Cells is Mediated in Part Through Autocrine VEGF Stimulation. Mol. Vis. 2014, 20, 781–789. [Google Scholar]
- Hobson, B.D.; Sulzer, D. Neuronal Presentation of Antigen and Its Possible Role in Parkinson’s Disease. J. Park. Dis. 2022, 12 (Suppl. S1), 137–147. [Google Scholar] [CrossRef]
- Siddiqui, A.; Hanson, I.; Andersen, J.K. Mao-B Elevation Decreases Parkin’s Ability to Efficiently Clear Damaged Mitochondria: Protective Effects of Rapamycin. Free Radic. Res. 2012, 46, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Probert, L. TNF and Its Receptors in the CNS: The Essential, the Desirable and the Deleterious Effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef]
- Hu, G.; Wang, Z.; Zhang, R.; Sun, W.; Chen, X. The Role of Apelin/Apelin Receptor in Energy Metabolism and Water Homeostasis: A Comprehensive Narrative Review. Front. Physiol. 2021, 12, 632886. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.N.; Stoyanov, D.S.; Pavlov, S.P.; Tonchev, A.B. Distribution, Function, and Expression of the Apelinergic System in the Healthy and Diseased Mammalian Brain. Genes 2022, 13, 2172. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhao, Q.; Chen, L.; Li, Z. Apelin/APJ System: An Emerging Therapeutic Target for Neurological Diseases. Mol. Biol. Rep. 2022, 50, 1639–1653. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Li, P.; Zheng, Y.; Yang, Y.; Ji, S. Apelin/APJ System in Inflammation. Int. Immunopharmacol. 2022, 109, 108822. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Song, J.; Xie, M.; Liu, Y.; Dong, Z.; Liu, X.; Li, X.; Zhang, M.; Chen, Y.; et al. Elabela Alleviates Ferroptosis, Myocardial Remodeling, Fibrosis and Heart Dysfunction in Hypertensive Mice by Modulating the IL-6/STAT3/GPX4 Signaling. Free Radic. Biol. Med. 2022, 181, 130–142. [Google Scholar] [CrossRef]
- Monastero, R.; Magro, D.O.; Venezia, M.; Pisano, C.; Balistreri, C.R. A Promising Therapeutic Peptide and Preventive/Diagnostic Biomarker for Age-Related Diseases: The Elabela/Apela/Toddler Peptide. Ageing Res. Rev. 2023, 91, 102076. [Google Scholar] [CrossRef]
- Dogantekin, A.; Dogantekin, E.; Gozel, N. The Neuronal Antiinflammatory Effects of Apelin-13. Turk. J. Health Sci. Life 2023, 6, 97–102. [Google Scholar] [CrossRef]
- Pope, G.R.; Tilve, S.; McArdle, C.A.; Lolait, S.J.; O’Carroll, A. Agonist-Induced Internalization and Desensitization of the Apelin Receptor. Mol. Cell. Endocrinol. 2016, 437, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Nakawaki, M.; Kenmoku, T.; Uchida, K.; Arendt-Nielsen, L.; Nagura, N.; Takaso, M. Expression of Apelin in Rotator Cuff Tears and Examination of Its Regulatory Mechanism: A Translational Study. Curēus 2023, 15, 44347. [Google Scholar] [CrossRef]
- Melgar-Lesmes, P.; Pauta, M.; Reichenbach, V.; Casals, G.; Ros, J.; Bataller, R.; Morales-Ruiz, M.; Jiménez, W. Hypoxia and Proinflammatory Factors Upregulate Apelin Receptor Expression in Human Stellate Cells and Hepatocytes. Gut 2011, 60, 1404–1411. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shan, B.; Liang, Y.; Wei, H.; Yuan, J. Parkin Regulates NF-ΚB by Mediating Site-Specific Ubiquitination of RIPK1. Cell Death Dis. 2018, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Casabona, J.C.; Puntel, M.; Pitossi, F.J. Interleukin-1β and Tumor Necrosis Factor-α: Reliable Targets for Protective Therapies in Parkinson’s Disease? Front. Cell. Neurosci. 2013, 7, 53. [Google Scholar] [CrossRef]
- Anilkumar, S.; Wright-Jin, E. NF-κB as an Inducible Regulator of Inflammation in the Central Nervous System. Cells 2024, 13, 485. [Google Scholar] [CrossRef]
- Kim, T.W.; Koo, S.Y.; Riessland, M.; Chaudhry, F.; Kolisnyk, B.; Cho, H.S.; Russo, M.V.; Saurat, N.; Mehta, S.; Garippa, R.; et al. TNF-NF-κB-p53 Axis Restricts In Vivo Survival of hPSC-derived Dopamine Neurons. Cell 2024, 187, 3671–3689.e23. [Google Scholar] [CrossRef]
- Shao, Z.Q.; Dou, S.S.; Zhu, J.G.; Wang, H.Q.; Wang, C.M.; Cheng, B.H.; Bai, B. Apelin-13 Inhibits Apoptosis and Excessive Autophagy in Cerebral Ischemia/Reperfusion Injury. Neural Regener. Res. 2021, 16, 1044–1051. [Google Scholar] [CrossRef]
- Day, R.T.; Cavaglieri, R.C.; Feliers, D. Apelin retards the progression of diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2013, 304, 788–800. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 14 February 2024).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H.G. Profiler-Interoperable Web Service for Functional Enrichment Analysis and Gene Identifier Mapping (2023 Update). Nucleic Acids Res. 2023, 51, gkad347. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Sergushichev, A. Fast Gene Set Enrichment Analysis. bioRxiv 2016. [Google Scholar] [CrossRef]
- Carbon, S.; Mungall, C. Gene Ontology Data Archive. Zenodo (CERN European Organization for Nuclear Research). 2023. Available online: https://zenodo.org/records/10162580 (accessed on 4 March 2024).
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein–Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Status | Donor | Differentiation Status | Genotype | iPSC Cell Line Symbol |
|---|---|---|---|---|
| HD | Healthy female, 18 years | Neurons | normal | IPSHD1.1S [20] |
| PD | PD female, disease onset 30 years, biopsy 41 years | Neurons | hom EX8 del PARK2 | IPSPDS13 [21] |
| PD | PD male, disease onset 38 years, biopsy 40 years | Neurons | het EX2 del PARK2 | IPSPDS14 [22] |
| HD | Healthy female, 26 years | Astrocytes | normal | IPSFD3.9L [20] |
| HD | Healthy female, 18 years | Astrocytes | normal | IPSHD1.1S [20] |
| PD | PD male, disease onset 40 years, biopsy 54 years | Astrocytes | het EX2 dup PARK2 | IPSPDS12 [23] |
| PD | PD male, disease onset 38 years, biopsy 40 years | Astrocytes | het EX2 del PARK2 | IPSPDS14 [22] |
| Comparison | HD Neurons | PD Neurons | ||||
|---|---|---|---|---|---|---|
| Up | Down | All | Up | Down | All | |
| HD glial mixed medium vs. base glial medium | 2057 | 2045 | 4102 | 510 | 632 | 1142 |
| PD glial mixed medium vs. base glial medium | 273 | 133 | 406 | 581 | 392 | 973 |
| PD glial mixed medium vs. HD glial mixed medium | 2111 | 1983 | 4094 | 521 | 296 | 817 |
| Short Comparison Name | Scheme of Bioinformatic Data Analysis | Comparison Meaning |
|---|---|---|
| PD0vsHD0 | (PD0vsHD0) vs. (PD1vsHD1) | The differences between PD and HD neurons in the base neuronal medium. |
| PD2vsHD2 | (PD2vsHD2) vs. (PD1vsHD1) | The differences in the influence of secretome of HD glia on PD and HD neurons. |
| PD3vsHD3 | (PD3vsHD3) vs. (PD1vsHD1) | The differences in the influence of secretome of PD glia on PD and HD neurons. |
| HD3vsHD2 | (HD3vsHD1) vs. (HD2vsHD1) | The differences in the influence of secretome of HD and PD glia on HD neurons. |
| PD3vsPD2 | (PD3vsPD1) vs. (PD2vsPD1) | The differences in the influence of secretome of HD and PD glia on PD neurons. |
| PD3vsHD2 | (PD3vsHD2) vs. (PD1vsHD1) | The differences between neurons in the “own system”: PD neurons in PD glial mixed medium and HD neurons in HD glial mixed medium. |
| Pathway ID | Pathway | PD0vsHD0 | PD3vsHD2 | PD2vsHD2 | HD3vsHD2 | PD3vsHD3 | PD3vsPD2 |
|---|---|---|---|---|---|---|---|
| GO:0034135 | regulation of Toll-like receptor 2 signaling pathway | enriched | |||||
| GO:0034140 | negative regulation of Toll-like receptor 3 signaling pathway | enriched | enriched | enriched | |||
| GO:0038061 | non-canonical NF-kappaB signal transduction | enriched | |||||
| GO:0000165 | MAPK cascade | enriched | enriched | ||||
| GO:0038066 | p38MAPK cascade | enriched | enriched | enriched | |||
| GO:0051896 | regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction | enriched | |||||
| GO:0035976 | transcription factor AP-1 complex | enriched | |||||
| GO:1990622 | CHOP-ATF3 complex | enriched | enriched | ||||
| GO:0036488 | CHOP-C/EBP complex | enriched | |||||
| GO:0038160 | CXCL12-activated CXCR4 signaling pathway | enriched | enriched | ||||
| GO:0070371 | ERK1 and ERK2 cascade | enriched | |||||
| GO:0006959 | humoral immune response | enriched | enriched | ||||
| GO:0019955 | cytokine binding | enriched | |||||
| GO:0004896 | cytokine receptor activity | enriched | |||||
| GO:0005125 | cytokine activity | enriched | enriched | enriched | |||
| GO:0019221 | cytokine-mediated signaling pathway | enriched | enriched | ||||
| GO:1990868 | response to chemokine | enriched | |||||
| GO:0034097 | response to cytokine | enriched | enriched | enriched | |||
| GO:0070555 | response to interleukin-1 | enriched | |||||
| GO:0034612 | response to tumor necrosis factor | enriched | |||||
| GO:0032611 | interleukin-1 beta production | enriched | |||||
| GO:0032613 | interleukin-10 production | enriched | |||||
| GO:0032623 | interleukin-2 production | enriched | |||||
| GO:0032633 | interleukin-4 production | enriched |
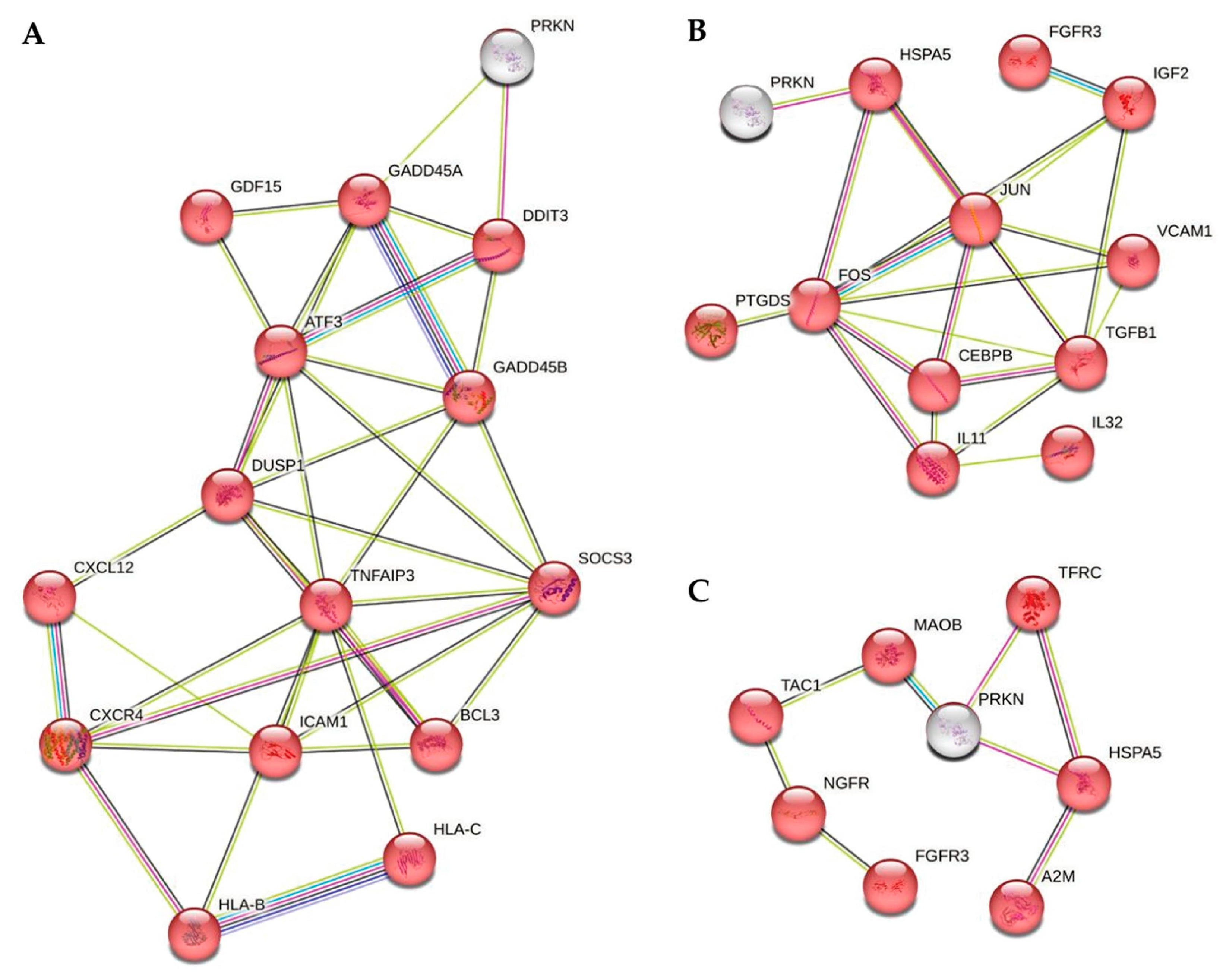
| Gene | Annotation | Influence on Neuroinflammation | Association with Neurodegeneration, PD and/or Parkin |
|---|---|---|---|
| DEGs associated with disease status of neurons | |||
| CXCL12/CXCR4 | C-X-C Motif Chemokine Ligand 12 | Cause MAPK1/MAPK3 activation and release of pro-inflammatory cytokines [25]. | Up-regulated in post-mortem brains of PD individuals [26]. |
| DDIT3 | DNA Damage Inducible Transcript 3 | Promotes NLRP3 inflammasome formation [27]. | NRLP3 is a substrate for ubiquitination by Parkin [28]. * DDIT3 was down-regulated in SH-SY5Y cells upon expression of the WT and the mutant Parkin compared to cells not expressing Parkin [29]. * |
| ICAM1 | Intracellular Adhesion Molecule 1 | Is a ligand for leucocyte adhesion protein [30]. | Suppressing ICAM-1 leads to a significant decrease in inflammation in MPTP PD mouse model [31]. |
| HLA-B HLA-C | Members of Major Histocompatibility Complex, Class I | Monitors the state of the organism by the immune system, presentation of mitochondrial antigens [32]. | DA neurons express MHC class I molecules in presence of activated microglia or other stressors linked to inflammation, becoming “visible” to the immune system; in the absence of Parkin, high levels of mitochondrial antigens are presented on MHC class I molecules in both macrophages and dendritic cells [32]. * |
| TNFAIP3 | TNF Alpha Induced Protein 3 | Terminates the NF-κB acting as ubiquitin-editing enzyme [33]. | Down-regulated in whole blood from PD patients [34]. Reduced in the SN of PD patients and mice and in the MPP+-treated SK-N-SH cells; plays a neuroprotective role in MPTP-induced mice by restricting NF-κB and mTOR pathways [33]. |
| SOCS3 | Suppressor of Cytokine Signaling 3 | Regulates a STAT3-mediated chemokine and chemokine receptor function, causes degradation of p65 NF-κB [35,36]. | In PD, SOCS signaling is inhibited, which leads to neuroinflammation and subsequent degeneration of DA neurons [37]. |
| DUSP1 | Dual Specificity Phosphatase 1 | Inactivates MAPKs by dephosphorylation [38]. | Up-regulated in PD [39]. LRRK2 was found to interact with DUSP1 and DUSP16 [40]. |
| ATF3 | Activating Transcription Factor 3 | Suppresses the expression of various inflammatory genes induced by TLR4 signaling, including genes encoding IL-6, IL-12b, and TNF-α [41]. | Up-regulated in midbrain neurons treated with 6-OHDA; neurons from ATF3 “knockout” mice were not protected from cell death [42]. |
| BCL3 | BCL3 Transcription Coactivator | Regulates the activity of NF-κB depending on the stimulation [43]. | Presented in the Hereditary Parkinsonism Gene Set [44]. |
| GDF15 | Growth Differentiation Factor 15 | Acts as an inflammatory protein, but also has protective and trophic functions for neurons [45]. | Promoted survival of DA neurons and regulated the inflammatory response after intrastriatal 6-OHDA lesion [46]. |
| GADD45A | Growth Arrest and DNA Damage Inducible Alpha | Is neuroprotective and can prevent neurodegeneration [47]. | The lack of Gadd45a gene expression exacerbates neuroinflammation in mice [47]. Up-regulated early after intrastriatal 6-OHDA lesion in rats specifically in substantia nigra DA neurons [48]. Parkin overexpression reduces GADD45A expression in ceramide-treated PC12 cells [49]. * |
| DEGs characteristic for PD neurons in HD glial mixed medium | |||
| JUNB/FOS | AP-1 Transcription Factor Subunits | Activate IL-6 and TNF-α production [50]. | AP-1 promotes the vicious cycle of neuroinflammation in microglia of the SNpc, driving DA neurons toward apoptosis [37]. |
| CEBPB | CCAAT Enhancer Binding Protein Beta | Acts as a positive regulator of cytokine gene expression [41]. | Depletion of C/EBPβ diminishes DA neuronal loss in human α-SNCA transgenic mice. C/EBPβ/AEP pathway correlates with Lewy body pathologies in human PD patients [51]. |
| VCAM1 | Vascular Cell Adhesion Molecule 1 | Promotes leukocyte adhesion and transmigration across the vascular endothelium; IL-1β and TNF-α up-regulated VCAM-1 [52]. | Levels of the soluble VLA4 ligand VCAM1 are highly increased in patients with PD [53]. In cultured human lung endothelial cells, down-regulation of Parkin by siRNA decreases LPS-induced VCAM-1 expression [54]. * |
| IGF2 | Insulin Like Growth Factor 2 | Reduces microgliosis, decreases the expression of pro-inflammatory cytokines [55]. | IGF2 plasma levels and IGF2 mRNA and protein levels are significantly decreased in PBMCs in PD patients; a relationship between IGF2 levels and the autophagy process in PD was found [56]. |
| TGFB1 | Transforming Growth Factor Beta 1 | Represses inflammatory response [57]. | Neuroprotective TGF-β signaling pathway is down-regulated during development of PD [37]. TGF-β1 expression is increased in biopsies of PD patients [58]. |
| FGFR3 | Fibroblast Growth Factor Receptor 3 | Activates MAPK and IL6 and IL8 production [59]. | Neuronal expression of FGFR3 is increased in post-mortem Lewy body disease brains [60]. |
| HSPA5 | Heat Shock Protein Family A (Hsp70) Member 5 | Performs quality control of proteins, suppresses α-synuclein aggregation and toxicity [61]. | GRP78/BIP/HSPA5 is potential target for the regulation of ER stress in experimental PD models [61]. |
| PTGDS | Prostaglandin D2 Synthase | Provides anti-inflammatory and antioxidant functions [62]. | Increased in twins with PD relative to healthy twins [63]. Is linked with neurodegenerative disease and brain injury; mediates anti-inflammatory effects of DJ-1, a PD-related gene [62]. |
| DEGs characteristic for HD neurons in PD glial mixed medium | |||
| NGFR (p75NTR) | Nerve Growth Factor Receptor | Activates NF-kB and MAPK [64]. | NGFR signaling pathway induces a double-sword effect, either detrimental or beneficial depending on the ligands and status of PD neuropathology [65]. |
| FGFR3 | Fibroblast Growth Factor Receptor 3 | Activates MAPK and IL6 and IL8 production [59]. | Neuronal expression of FGFR3 was increased in post-mortem Lewy body disease brains [60]. |
| TAC1 | Tachykinin Precursor 1 | Is responsible for the high density of microglia, up-regulates inflammation [66]. | SP encoded by TAC1 gene exacerbated DA cell death; in the 6-OHDA-induced model of PD, the administration of additional SP accelerated disease progression [66]. |
| TFRC | Transferrin Receptor | Is the primary iron import mechanism; induces α-synuclein fibril formation by ROS production [67]. | Strongly up-regulated in TPFPD-treated (a chronic-type inflammatory stimulation combining PD patient-derived αSYN fibrils, TNF-α and prostaglandin E2) human induced-microglial-like cells [67]. |
| MAOB | Monoamine Oxidase B | Is associated with reactive astrogliosis [68]. | Comprehensively reviewed MAOB’s role as a DA-degrading enzyme and as a critical enzyme regulating astrocytic reactivity [68]. Stable transfection of Parkin in the human DA neuroblastoma cell line SH-SY5Y markedly reduced the activities of MAOB [69]. * |
| A2M | Alpha-2-Macroglobulin | Binds pro-inflammatory cytokines, cytokine clearance [70]. | Increased plasma A2M level in AD patients; A2M participated in delay of PD neuroinflammation [71]. |
| HSPA5 | Heat Shock Protein Family A (Hsp70) Member 5 | Performs quality control of proteins, suppresses α-synuclein aggregation and toxicity [61]. | GRP78/BIP/HSPA5 is potential target for the regulation of ER stress in experimental PD models [61]. |
| Marker | Antibodies | Dilution | Company, Cat # |
|---|---|---|---|
| Primary antibodies | anti-TUBB3 | 1:2000 | Abcam, #AB7751 |
| Rabbit anti-S100 | 1:2 | Agilent Dako, #GA50461-2 | |
| Secondary antibodies | Goat anti-Rabbit IgG (H + L), AF546 | 1:1000 | TermoFisher, #A11010 |
| Goat anti-Mouse IgG (H + L), AF488 | 1:1000 | TermoFisher, #A11008 |
| Gene | Forward | Reverse | Annealing t °C |
|---|---|---|---|
| TNFAIP3 | CTGAAAACGAACGGTGACGG | CCATGGGTGTGTCTGTGGAA | 60 |
| DDIT3 | CACCTCCTGGAAATGAAGAGGAAG | CTCTGGGAGGTCTTGTGAC | 60 |
| GADD45A | GAATTCTCGGCTGGAGAGCA | ACGTTATCGGGGTCGACGTT | 65 |
| ATF3 | CTCTGCGCTGGAATCAGTCA | CTTCTTCAGGGGCTACCTCG | 60 |
| NGFR | TGCCAGGACAAGCAGAACAC | CCAGGGATCTCCTCGCAC | 60 |
| FGFR3 | CTGGGAGATCTTCACGCTGG | GTCCAGGTACTCGTCGGTG | 60 |
| A2M | AGCAGGAAGACATGAAGGGC | AATCACGTCCCCGGTAGGTA | 60 |
| HSPA5 | CATCAACGAGCCTACGGCA | AGACACATCGAAGGTTCCGC | 65 |
| APLNR | GTCAGCCAGCTACTCTTCGG | AGG GGATGGATTTCTCGTGC | 60 |
| APLN | GCTGGAAGACGGCAATGTCC | GCGGAATTTCCTCCGACCT | 60 |
| TUBB3 | CTCAGGGGCCTTTGGACATC | CAGGCAGTCGCAGTTTTCAC | 60 |
| NEUN | TACGCAGCCTACAGATACGCTC | TGGTTCCAATGCTGTAGGTCGC | 60 |
| AADC | CTCGGACCAAAGTGATCCAT | GGGTGGCAACCATAAAGAAA | 60 |
| MAP2 | AGGCTGTAGCAGTCCTGAAAGG | CTTCCTCCACTGTGACAGTCTG | 60 |
| GLUT1 | GCTGTGCTTATGGGCTTCTC | CACATACATGGGCACAAAGC | 60 |
| VIM | ATTCCACTTTGCGTTCAAGG | CTTCAGAGAGAGGAAGCCGA | 60 |
| TNRSF1B | CACCGGGAGCTCAGATTCTT | CCGAAAGGCACATTCCTCCT | 60 |
| 18S | CGGCTACCACATCCAAGGAA | GCTGGAATTACCGCGGCT | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerasimova, T.; Poberezhniy, D.; Nenasheva, V.; Stepanenko, E.; Arsenyeva, E.; Novosadova, L.; Grivennikov, I.; Illarioshkin, S.; Lagarkova, M.; Tarantul, V.; et al. Inflammatory Intracellular Signaling in Neurons Is Influenced by Glial Soluble Factors in iPSC-Based Cell Model of PARK2-Associated Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 9621. https://doi.org/10.3390/ijms25179621
Gerasimova T, Poberezhniy D, Nenasheva V, Stepanenko E, Arsenyeva E, Novosadova L, Grivennikov I, Illarioshkin S, Lagarkova M, Tarantul V, et al. Inflammatory Intracellular Signaling in Neurons Is Influenced by Glial Soluble Factors in iPSC-Based Cell Model of PARK2-Associated Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(17):9621. https://doi.org/10.3390/ijms25179621
Chicago/Turabian StyleGerasimova, Tatiana, Daniil Poberezhniy, Valentina Nenasheva, Ekaterina Stepanenko, Elena Arsenyeva, Lyudmila Novosadova, Igor Grivennikov, Sergey Illarioshkin, Maria Lagarkova, Vyacheslav Tarantul, and et al. 2024. "Inflammatory Intracellular Signaling in Neurons Is Influenced by Glial Soluble Factors in iPSC-Based Cell Model of PARK2-Associated Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 17: 9621. https://doi.org/10.3390/ijms25179621
APA StyleGerasimova, T., Poberezhniy, D., Nenasheva, V., Stepanenko, E., Arsenyeva, E., Novosadova, L., Grivennikov, I., Illarioshkin, S., Lagarkova, M., Tarantul, V., & Novosadova, E. (2024). Inflammatory Intracellular Signaling in Neurons Is Influenced by Glial Soluble Factors in iPSC-Based Cell Model of PARK2-Associated Parkinson’s Disease. International Journal of Molecular Sciences, 25(17), 9621. https://doi.org/10.3390/ijms25179621