
Discovery of Indole–Thiourea Derivatives as Tyrosinase Inhibitors: Synthesis, Biological Evaluation, Kinetic Studies, and In Silico Analysis



Abstract
:1. Introduction
2. Results and Discussion
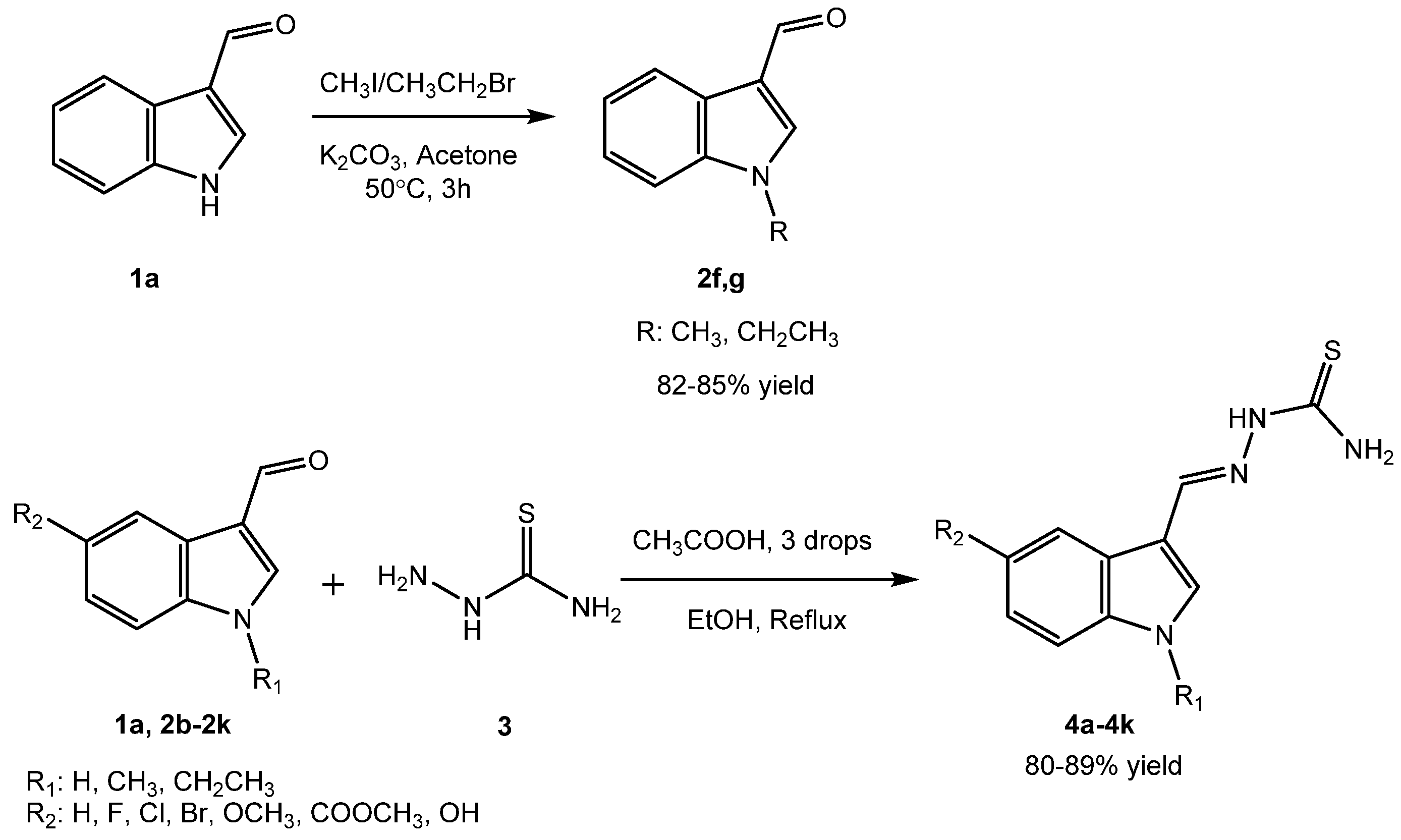
2.1. Chemistry
2.2. Biological Evaluations
2.2.1. Anti-Tyrosinase Activity and Structure–Activity Relationship (SAR)
2.2.2. Determination of the Inhibitory Mechanism through Enzyme Kinetics
2.2.3. Free Radical-Scavenging Activity
2.3. In Silico Experiments
2.3.1. Molecular Properties and Drug Likeness
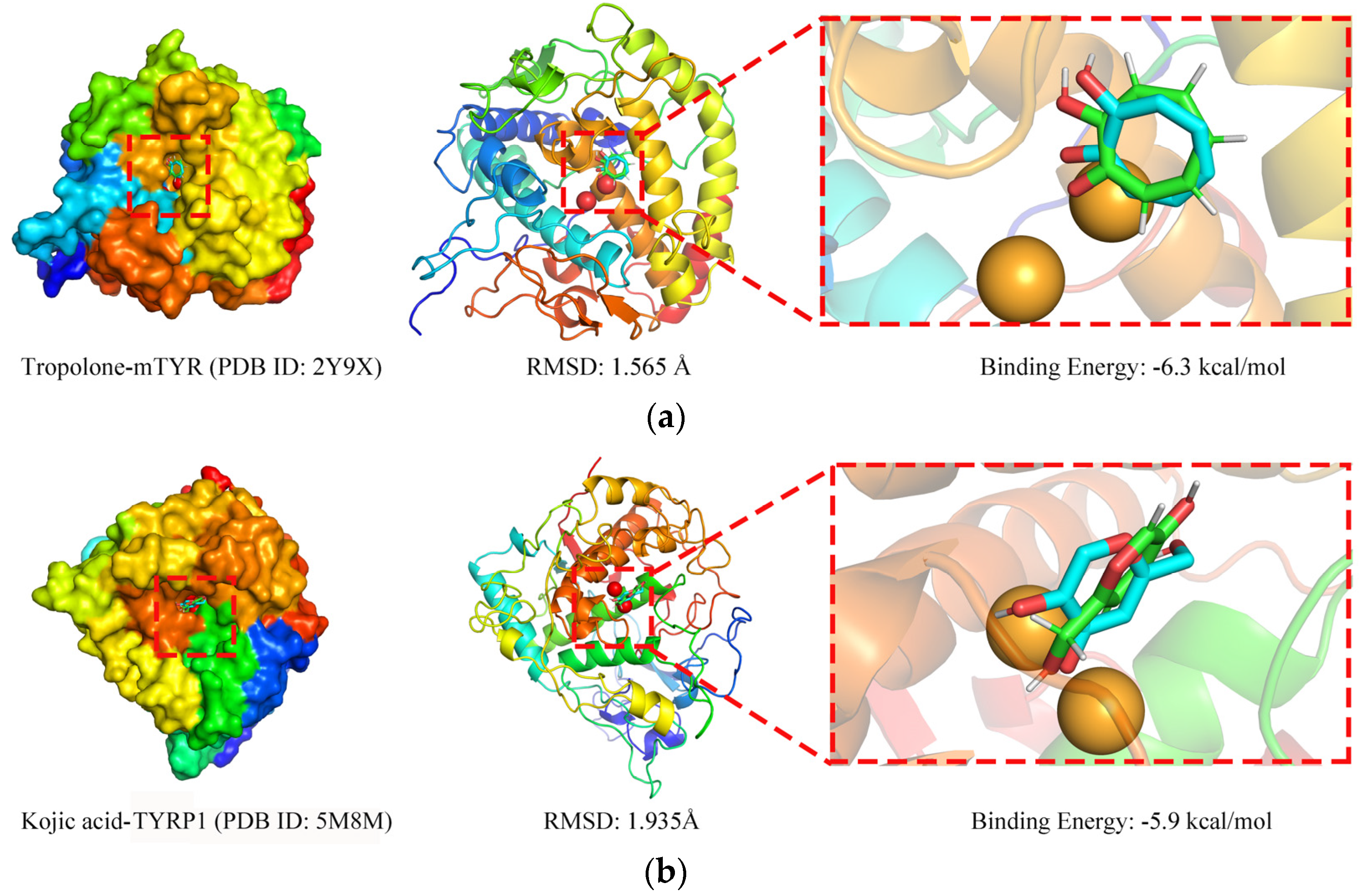
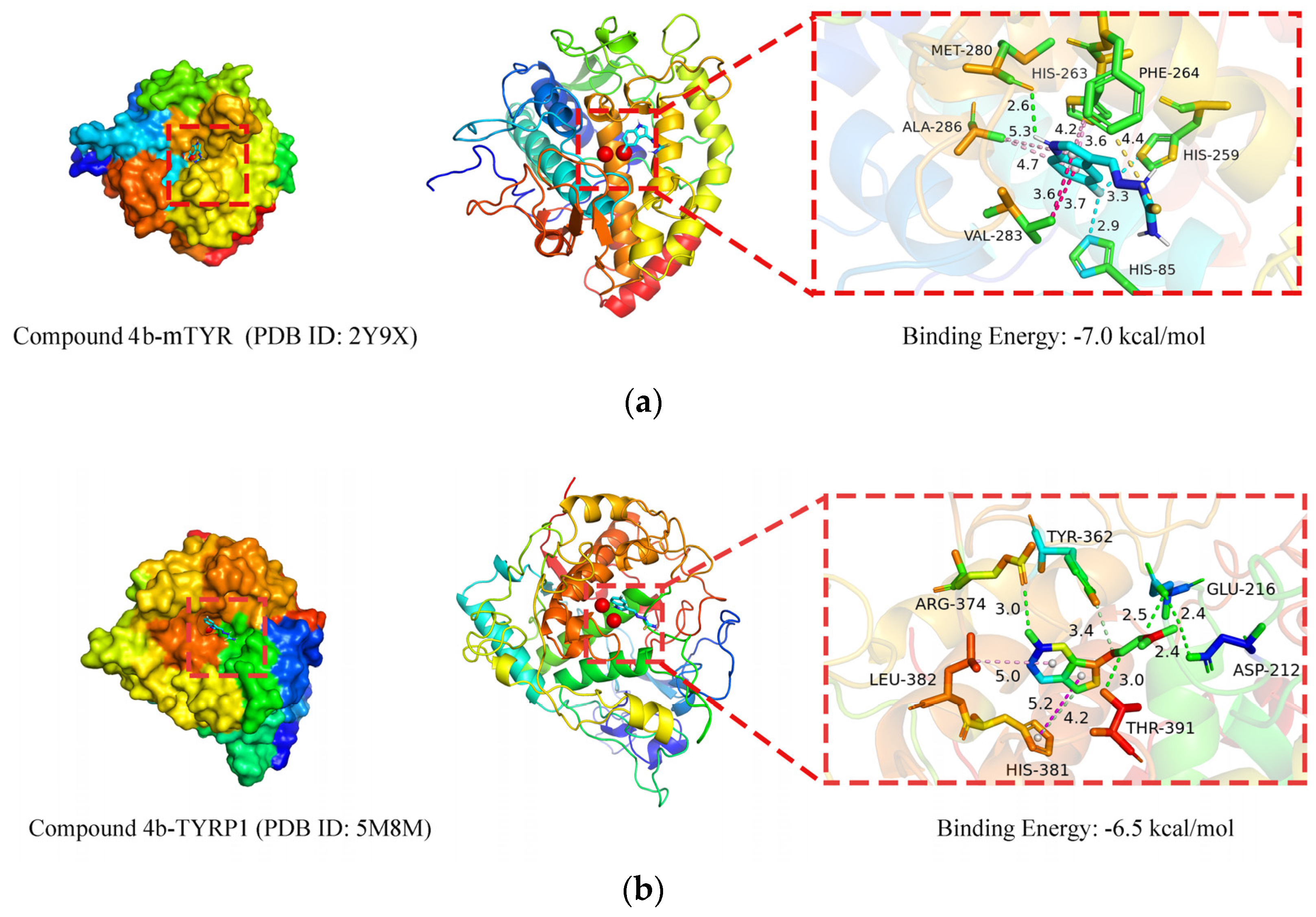
2.3.2. Molecular Docking Simulation
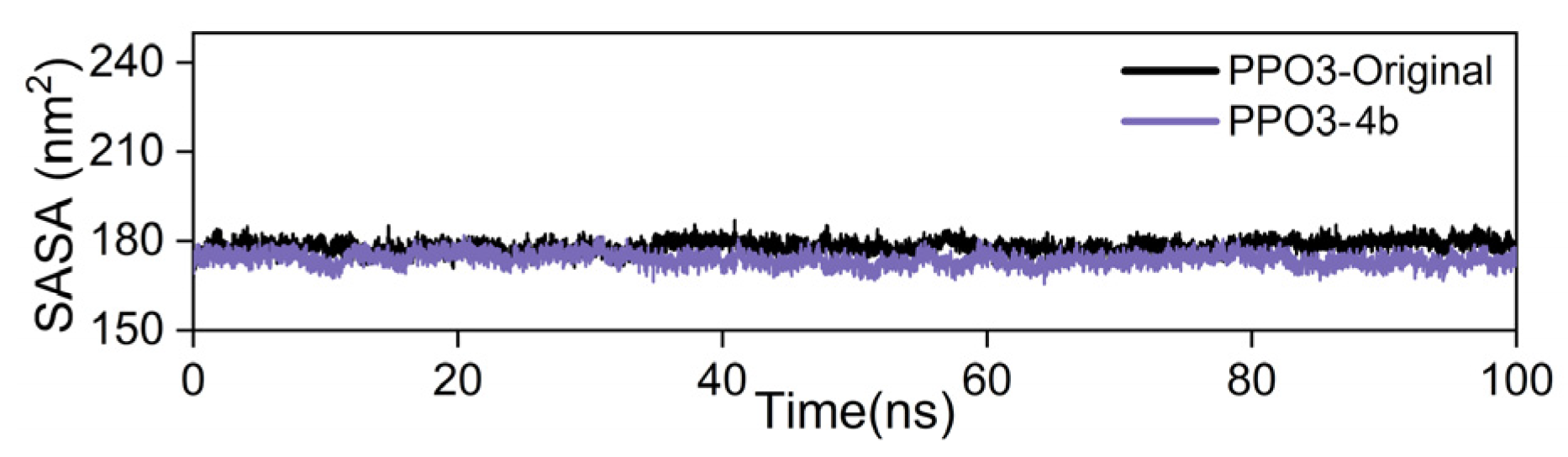
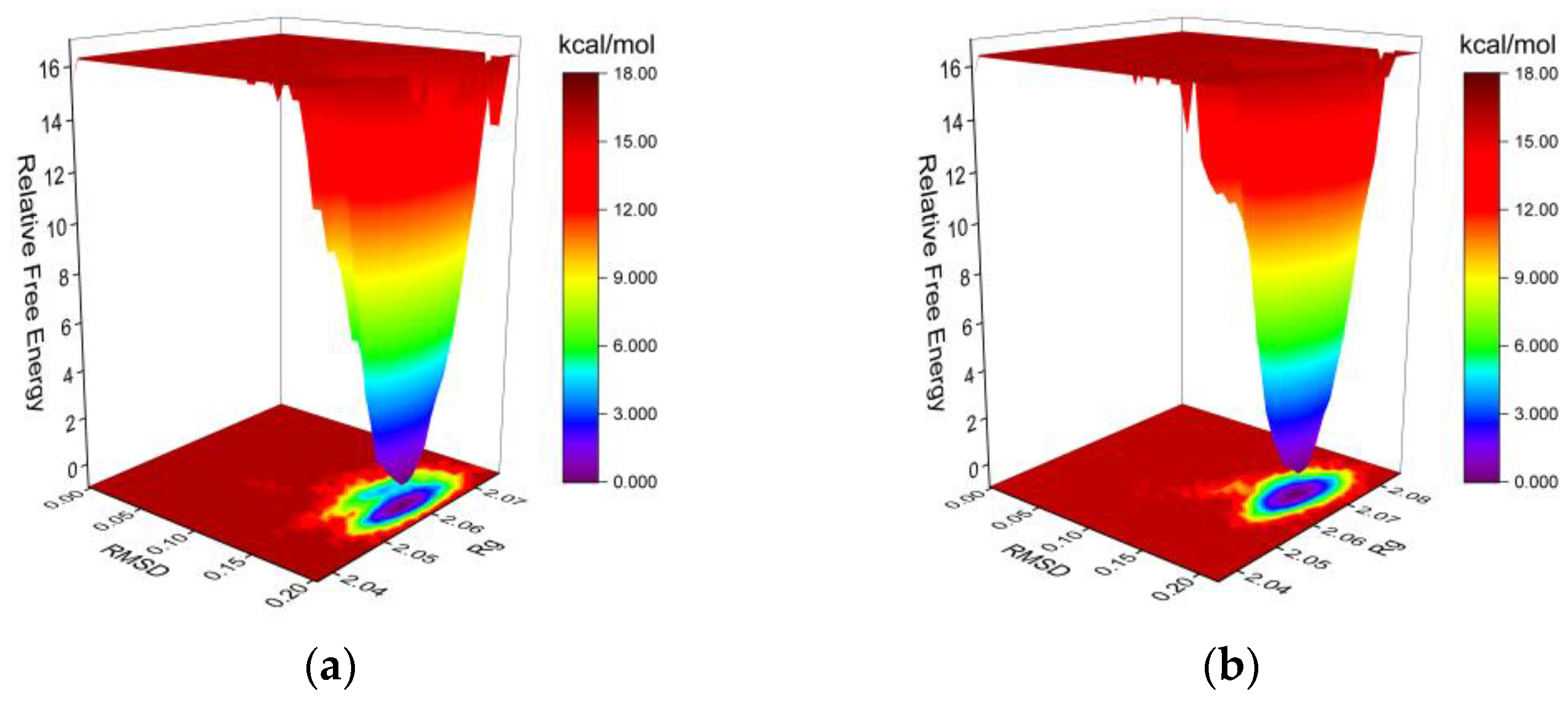
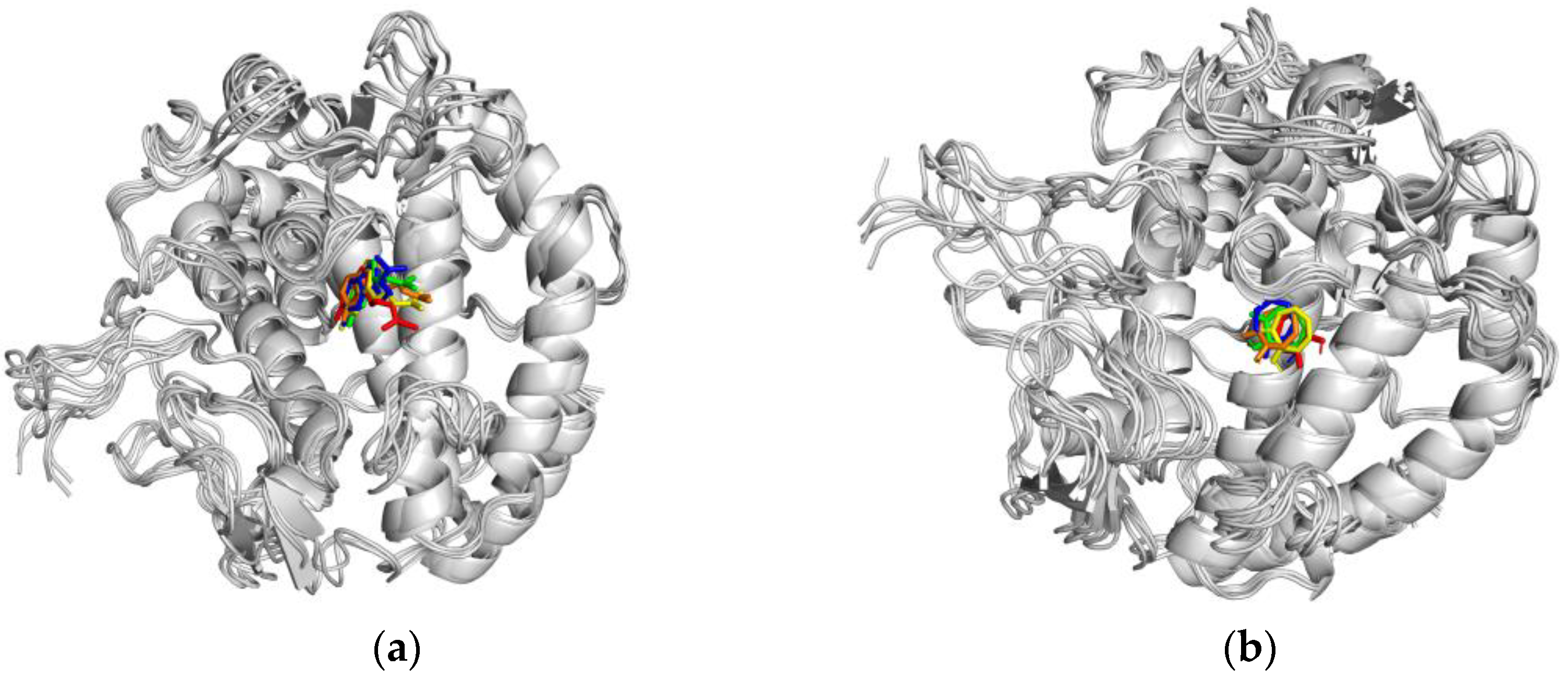
2.3.3. Molecular Dynamics (MD) Simulation
3. Materials and Methods
3.1. Chemicals and Instruments
3.2. General Procedure for the Synthesis of the Compounds
3.2.1. Procedure for the Synthesis of Indole-3-Carbaldehyde Derivatives (2f, g)
- 1-ethyl-1H-indole-3-carbaldehyde (2f)
- 1-methyl-1H-indole-3-carbaldehyde (2g)
3.2.2. Procedure for the Synthesis of Indole–Thiourea Derivatives (4a–4k)
- (E)-2-((1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4a)
- (E)-2-((5-fluoro-1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4b)
- (E)-2-((5-chloro-1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4c)
- (E)-2-((5-bromo-1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4d)
- (E)-2-((5-methoxy-1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4e)
- (E)-2-((1-ethyl-1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4f)
- (E)-2-((1-methyl-1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4g)
- (E)-2-((5-methoxy-1-methyl-1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4h)
- (E)-2-((5-bromo-1-methyl-1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4i)
- Methyl(E)-3-((2-carbamothioylhydrazineylidene)methyl)-1H-indole-5-carboxylate (4j)
- (E)-2-((5-hydroxy-1H-indol-3-yl)methylene)hydrazine-1-carbothioamide (4k)
3.3. Biology
3.3.1. Tyrosinase Inhibition Assay
3.3.2. Kinetic Mechanism Analysis
3.3.3. Free Radical-Scavenging Assay
3.4. Computational Methodology
3.4.1. Molecular Properties and Drug Likeness
3.4.2. Molecular Docking Simulation
3.4.3. Molecular Dynamics (MD) Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, Z.; Wang, G.; Zeng, Q.H. A systematic review of synthetic tyrosinase inhibitors and their structure-activity relationship. Crit. Rev. Food Sci. Nutr. 2022, 62, 4053–4094. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Lin, L.C.; Yang, W.F. An updated organic classification of tyrosinase inhibitors on melanin biosynthesis. Curr. Org. Chem. 2015, 19, 4–18. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Hearing, V.J. Physiological factors that regulate skin pigmentation. Biofactors 2009, 35, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Thawabteh, A.M.; Jibreen, A.; Karaman, D. Skin pigmentation types, causes and treatment—A review. Molecules 2023, 28, 4839. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Hassan, K.M.T. A comprehensive review on tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef]
- Panzella, L.; Napolitano, A. Natural and bioinspired phenolic compounds as tyrosinase inhibitors for the treatment of skin hyperpigmentation: Recent advances. Cosmetics 2019, 6, 57. [Google Scholar] [CrossRef]
- Masum, M.N.; Yamauchi, K.; Mitsunaga, T. Tyrosinase inhibitors from natural and synthetic sources as skin-lightening agents. Rev. Agric. Sci. 2019, 7, 41–58. [Google Scholar] [CrossRef]
- Solano, F.; Briganti, S.; Picardo, M. Hypopigmenting agents: An updated review on biological, chemical and clinical aspects. Pigment. Cell Res. 2006, 19, 550–571. [Google Scholar] [CrossRef]
- Couteau, C.; Coiffard, L. Overview of skin whitening agents: Drugs and cosmetic products. Cosmetics 2016, 3, 27. [Google Scholar] [CrossRef]
- Chai, L.; Zhou, J.; Feng, H. Functionalized carbon quantum dots with dopamine for tyrosinase activity monitoring and inhibitor screening: In vitro and intracellular investigation. ACS Appl. Mater. Interfaces 2015, 7, 23564–23574. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Evans, J.; Ito, S. Nonenzymatic spontaneous oxidative transformation of 5,6-dihydroxyindole. Int. J. Mol. Sci. 2020, 21, 7321. [Google Scholar] [CrossRef]
- Dolinska, M.B.; Sergeev, Y.V. Molecular Modeling of the Multiple-Substrate Activity of the Human Recombinant Intra-Melanosomal Domain of Tyrosinase and Its OCA1B-Related Mutant Variant P406L. Int. J. Mol. Sci. 2024, 25, 3373. [Google Scholar] [CrossRef]
- Siddique, S.; Ahmad, K.R.; Nawaz, S.K. Evaluation of the anti-inflammatory, analgesic, anti-pyretic and anti-ulcerogenic potentials of synthetic indole derivatives. Sci. Rep. 2023, 13, 8639. [Google Scholar] [CrossRef] [PubMed]
- Jacob, Í.T.T.; Gomes, F.O.S. Anti-inflammatory activity of novel thiosemicarbazone compounds indole-based as COX inhibitors. Pharmacol. Rep. 2021, 73, 907–925. [Google Scholar] [CrossRef]
- Demurtas, M.; Baldisserotto, A.; Lampronti, I. Indole derivatives as multifunctional drugs: Synthesis and evaluation of antioxidant, photoprotective and antiproliferative activity of indole hydrazones. Bioorganic Chem. 2019, 85, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Konus, M.; Çetin, D.; Kızılkan, N.D. Synthesis and biological activity of new indole based derivatives as potent anticancer, antioxidant and antimicrobial agents. J. Mol. Struct. 2022, 1263, 133168. [Google Scholar] [CrossRef]
- Dhiman, A.; Sharma, R.; Singh, R.K. Target-based anticancer indole derivatives and insight into structure—activity relationship: A mechanistic review update (2018–2021). Acta Pharm. Sin. B 2022, 12, 3006–3027. [Google Scholar] [CrossRef]
- Devi, N.; Kaur, K.; Biharee, A. Recent development in indole derivatives as anticancer agent: A mechanistic approach. Anti-Cancer Agents Med. Chem. 2021, 21, 1802–1824. [Google Scholar] [CrossRef]
- Ferro, S.; Certo, G.; De, L.L. Searching for indole derivatives as potential mushroom tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31, 398–403. [Google Scholar] [CrossRef]
- Lu, L.; Hu, C.; Min, X. In vitro and in vivo biological evaluation of indole-thiazolidine-2, 4-dione derivatives as tyrosinase inhibitors. Molecules 2023, 28, 7470. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Geng, X.; Hashiguchi, M. Indole-3-carbaldehyde: A tyrosinase inhibitor from fungus YL185. J. Wood Sci. 2003, 49, 349–354. [Google Scholar] [CrossRef]
- Xu, Y.; Liang, X.; Hyun, C.G. Isolation, Characterization, Genome Annotation, and Evaluation of Tyrosinase Inhibitory Activity in Secondary Metabolites of Paenibacillus sp. JNUCC32: A Comprehensive Analysis through Molecular Docking and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2024, 25, 2213. [Google Scholar] [CrossRef] [PubMed]
- Melong, R.; Dzoyem, J.P.; Tsamo, A.T. Inhibitory effects of four naturally occurring compounds from Epicoccum nigrum on acetylcholinesterase activity and nitric oxide production in LPS-activated RAW 264.7 cells. Invest. Med. Chem. Pharmacol. 2018, 1, 18. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, M.K.; Kwon, S.B. Tumor apoptosis by indole-3-acetic acid/light in B16F10 melanoma-implanted nude mice. Arch. Dermatol. Res. 2009, 301, 319–322. [Google Scholar] [CrossRef]
- Kudličková, Z.; Michalková, R.; Salayová, A. Design, synthesis, and evaluation of novel indole hybrid chalcones and their antiproliferative and antioxidant activity. Molecules 2023, 28, 6583. [Google Scholar] [CrossRef]
- Citarella, A.; Moi, D.; Pedrini, M. Synthesis of SARS-CoV-2 M pro inhibitors bearing a cinnamic ester warhead with in vitro activity against human coronaviruses. Org. Biomol. Chem. 2023, 21, 3811–3824. [Google Scholar] [CrossRef]
- Garrepalli, S.; Gudipati, R.; Kapavarapu, R. Synthesis and characterization of two known and one new impurities of dolutegravir: In silico evaluation of certain intermediates against SARS-CoV-2 O-ribose methyltransferase (OMTase). J. Mol. Struct. 2023, 1271, 133992. [Google Scholar] [CrossRef]
- Iraji, A.; Sheikhi, N.; Attarroshan, M. Design, synthesis, spectroscopic characterization, in vitro tyrosinase inhibition, antioxidant evaluation, in silico and kinetic studies of substituted indole-carbohydrazides. Bioorganic Chem. 2022, 129, 106140. [Google Scholar] [CrossRef]
- Liu, J.; Yi, W.; Wan, Y. 1-(1-Arylethylidene) thiosemicarbazide derivatives: A new class of tyrosinase inhibitors. Bioorganic Med. Chem. 2008, 16, 1096–1102. [Google Scholar] [CrossRef]
- You, A.; Zhou, J.; Song, S. Rational design, synthesis and structure–activity relationships of 4-alkoxy-and 4-acyloxy-phenylethylenethiosemicarbazone analogues as novel tyrosinase inhibitors. Bioorganic Med. Chem. 2015, 23, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Hałdys, K.; Latajka, R. Thiosemicarbazones with tyrosinase inhibitory activity. MedChemComm 2019, 10, 378–389. [Google Scholar] [CrossRef]
- Dong, H.; Liu, J.; Liu, X. Molecular docking and QSAR analyses of aromatic heterocycle thiosemicarbazone analogues for finding novel tyrosinase inhibitors. Bioorganic Chem. 2017, 75, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Hałdys, K.; Goldeman, W.; Jewgiński, M. Inhibitory properties of aromatic thiosemicarbazones on mushroom tyrosinase: Synthesis, kinetic studies, molecular docking and effectiveness in melanogenesis inhibition. Bioorganic Chem. 2018, 81, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Riley, P.A. Melanin. Int. J. Biochem. Cell Biol. 1997, 29, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Manga, P.; Sato, K.; Ye, L. Mutational analysis of the modulation of tyrosinase by tyrosinase-related proteins 1 and 2 in vitro. Pigment Cell Res. 2000, 13, 364–374. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 403–425. [Google Scholar] [CrossRef]
- Mann, T.; Gerwat, W.; Batzer, J. Inhibition of human tyrosinase requires molecular motifs distinctively different from mushroom tyrosinase. J. Investig. Dermatol. 2018, 138, 1601–1608. [Google Scholar] [CrossRef]
- Lai, X.; Soler-Lopez, M.; Wichers, H.J. Large-scale recombinant expression and purification of human tyrosinase suitable for structural studies. PLoS ONE 2016, 11, 0161697. [Google Scholar] [CrossRef]
- You, Z.; WANG, Y.; HE, Q. Homology Modeling of Human Tyrosinase. J. Southeast Univ. Nat. Sci. Ed. 2020, 42, 42–48. [Google Scholar]
- Kim, H.J.; Jung, H.J.; Kim, Y.E. Investigation of the Efficacy of Benzylidene-3-methyl-2-thioxothiazolidin-4-one Analogs with Antioxidant Activities on the Inhibition of Mushroom and Mammal Tyrosinases. Molecules 2024, 29, 2887. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Yoshimori, A.; Ogawa, H. The structural differences between mushroom and human tyrosinase cleared by investigating the inhibitory activities of stilbenes. J. Mol. Struct. 2023, 1272, 134180. [Google Scholar] [CrossRef]
- Daungkamon, N.; Lalida, S.; Vannajan, S.L.; Piyarat, N. Estimation of Inhibitory Effect against Tyrosinase Activity through Homology Modeling and Molecular Docking. Enzym. Res. 2015, 2015, 12. [Google Scholar]
- Lima, C.R.; Silva, J.R.A. Combined kinetic studies and computational analysis on kojic acid analogs as tyrosinase inhibitors. Molecules 2014, 19, 9591–9605. [Google Scholar] [CrossRef]
- Wen, K.C.; Chang, C.S.; Chien, Y.C. Tyrosol and its analogues inhibit alpha-melanocyte-stimulating hormone induced melanogenesis. Int. J. Mol. Sci. 2013, 14, 23420–23440. [Google Scholar] [CrossRef]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M. Structure of human tyrosinase related protein 1 reveals a binuclear zinc active site important for melanogenesis. Angew. Chem. Int. Ed. 2017, 56, 9812–9815. [Google Scholar] [CrossRef]
- Liu, S.; Tian, M.; Bu, X. Covalent organic frameworks toward diverse photocatalytic aerobic oxidations. Chem.–A Eur. J. 2021, 27, 7738–7744. [Google Scholar] [CrossRef] [PubMed]
- Layer, R.W. The Chemistry of Imines. Chem. Rev. 1963, 63, 489–510. [Google Scholar] [CrossRef]
- Kovacic, P.; Somanathan, R. Mechanism of conjugated imine and iminium species, including marine alkaloids: Electron transfer, reactive oxygen species, therapeutics and toxicity. Curr. Bioact. Compd. 2010, 6, 46–59. [Google Scholar] [CrossRef]
- Kalia, J.; Raines, R.T. Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int. Ed. 2008, 47, 7523–7526. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; Bakr, R.B. Pyrimidine and fused pyrimidine derivatives as promising protein kinase inhibitors for cancer treatment. Med. Chem. Res. 2021, 30, 31–49. [Google Scholar] [CrossRef]
- Wang, L.; Chambers, J.; Abel, R. Protein–ligand binding free energy calculations with FEP+. In Biomolecular Simulations: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2019; pp. 201–232. [Google Scholar]
- Kalid, O.; Toledo, W.D.; Shechter, S. Consensus Induced Fit Docking (cIFD): Methodology, validation, and application to the discovery of novel Crm1 inhibitors. J. Comput. Aided Mol. Des. 2012, 26, 1217–1228. [Google Scholar] [CrossRef]
- Arai, M. Unified understanding of folding and binding mechanisms of globular and intrinsically disordered proteins. Biophys. Rev. 2018, 10, 163–181. [Google Scholar] [CrossRef]
- Gulcin, İ.; Alwasel, S.H. DPPH radical scavenging assay. Processes 2023, 11, 2248. [Google Scholar] [CrossRef]
- Minatel, I.O.; Borges, C.V.; Ferreira, M.I. Phenolic compounds: Functional properties, impact of processing and bioavailability. Phenolic Compd. Biol. Act. 2017, 8, 1–24. [Google Scholar]
- Merecz-Sadowska, A.; Sitarek, P.; Kowalczyk, T. Food anthocyanins: Malvidin and its glycosides as promising antioxidant and anti-inflammatory agents with potential health benefits. Nutrients 2023, 15, 3016. [Google Scholar] [CrossRef] [PubMed]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx 2005, 2, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.C.H.; Abagyan, R.; Totrov, M. Ligand-biased ensemble receptor docking (LigBEnD): A hybrid ligand/receptor structure-based approach. J. Comput. Aided Mol. Des. 2018, 32, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [PubMed]
- Zafrani, Y.; Parvari, G.; Amir, D. Modulation of the H-bond basicity of functional groups by α-fluorine-containing functions and its implications for lipophilicity and bioisosterism. J. Med. Chem. 2021, 64, 4516–4531. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Yamazaki, T.; Taguchi, T.; Ojima, I. Unique properties of fluorine and their relevance to medicinal chemistry and chemical biology. In Fluorine in Medicinal Chemistry and Chemical Biology; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2009; p. 1. [Google Scholar]
- Seo, S.; Choi, J.; Park, S. Binding affinity prediction for protein–ligand complex using deep attention mechanism based on intermolecular interactions. BMC Bioinform. 2021, 22, 1–15. [Google Scholar]
- Wang, D.D.; Wu, W.; Wang, R. Structure-based, deep-learning models for protein-ligand binding affinity prediction. J. Cheminformatics 2024, 16, 2. [Google Scholar] [CrossRef]
- Gioia, D.; Bertazzo, M.; Recanatini, M. Dynamic docking: A paradigm shift in computational drug discovery. Molecules 2017, 22, 2029. [Google Scholar] [CrossRef]
- Borgne, M.L.; Marchand, P.; Duflos, M. Synthesis and In Vitro Evaluation of 3-(1-Azolylmethyl)-1H-indoles and 3-(1-Azolyl-1-phenylmethyl)-1H-indoles as Inhibitors of P450 arom. Arch. Pharm. 1997, 330, 141–145. [Google Scholar] [CrossRef]
- Zheng, J.; Deng, L.; Chen, M. Elaboration of thorough simplified vinca alkaloids as antimitotic agents based on pharmacophore similarity. Eur. J. Med. Chem. 2013, 65, 158–167. [Google Scholar] [CrossRef]
- Haribabu, J.; Sabapathi, G.; Tamizh, M.M. Water-soluble mono-and binuclear Ru (η6-p-cymene) complexes containing indole thiosemicarbazones: Synthesis, DFT modeling, biomolecular interactions, and in vitro anticancer activity through apoptosis. Organometallics 2018, 37, 1242–1257. [Google Scholar] [CrossRef]
- Mashayekhi, V.; Tehrani, K.H.M.E.; Amidi, S. Synthesis of novel indole hydrazone derivatives and evaluation of their antiplatelet aggregation activity. Chem. Pharm. Bull. 2013, 61, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Dubois, C.; Yahiaoui, S. Refinement of arylthiosemicarbazone pharmacophore in inhibition of mushroom tyrosinase. Eur. J. Med. Chem. 2011, 46, 4330–4335. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Shi, S.; Yi, J. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 2024, 236, gkae236. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| No. | Compound | R1 | R2 | Tyrosinase Activity IC50 ± SEM (μM) |
| 1 | 4a | H | H | 7.9 ± 4.07 |
| 2 | 4b | H | F | 5.9 ± 2.47 |
| 3 | 4c | H | Cl | 13.2 ± 1.68 |
| 4 | 4d | H | Br | 14.9 ± 2.86 |
| 5 | 4e | H | OCH3 | 30.1 ± 1.77 |
| 6 | 4f | CH2CH3 | H | 126.6 ± 2.85 |
| 7 | 4g | CH3 | H | 155.5 ± 1.07 |
| 8 | 4h | CH3 | OCH3 | 163.3 ± 3.81 |
| 9 | 4i | CH3 | Br | 46.3 ± 3.11 |
| 10 | 4j | H | COOCH3 | 103.1 ± 2.72 |
| 11 | 4k | H | OH | 43.30 ± 3.46 |
| 12 | Kojic acid | 16.4 ± 3.53 | ||
| Compounds | SC50 ± SD (μM) |
|---|---|
| 4a | 482.7 ± 1.3 |
| 4b | 544.3 ± 1.5 |
| 4c | 659.2 ± 0.5 |
| 4d | 769.0 ± 2.6 |
| 4e | 603.4 ± 1.0 |
| 4f | >1000 |
| 4g | 493.5 ± 0.7 |
| 4h | 340.2 ± 3.1 |
| 4i | >1000 |
| 4j | 275.7 ± 2.7 |
| 4k | 24.6 ± 3.7 |
| Ascorbic acid | 37.9 ± 0.3 |
| Compounds | Binding Energy (kcal/mol) | |
|---|---|---|
| mTYR | TYRP1 | |
| 4a | −7.0 | −5.8 |
| 4b | −7.0 | −6.5 |
| 4c | −5.3 | −5.5 |
| 4d | −5.9 | −6.2 |
| 4e | −6.1 | −5.6 |
| 4f | −6.1 | −6.5 |
| 4g | −5.8 | −6.1 |
| 4h | −6.1 | −6.2 |
| 4i | −6.0 | −4.5 |
| 4j | −6.1 | −5.9 |
| 4k | −6.4 | −6.4 |
| Tropolone | −6.3 | / |
| Kojic acid | −5.3 | −5.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Liang, X.; Hyun, C.-G. Discovery of Indole–Thiourea Derivatives as Tyrosinase Inhibitors: Synthesis, Biological Evaluation, Kinetic Studies, and In Silico Analysis. Int. J. Mol. Sci. 2024, 25, 9636. https://doi.org/10.3390/ijms25179636
Xu Y, Liang X, Hyun C-G. Discovery of Indole–Thiourea Derivatives as Tyrosinase Inhibitors: Synthesis, Biological Evaluation, Kinetic Studies, and In Silico Analysis. International Journal of Molecular Sciences. 2024; 25(17):9636. https://doi.org/10.3390/ijms25179636
Chicago/Turabian StyleXu, Yang, Xuhui Liang, and Chang-Gu Hyun. 2024. "Discovery of Indole–Thiourea Derivatives as Tyrosinase Inhibitors: Synthesis, Biological Evaluation, Kinetic Studies, and In Silico Analysis" International Journal of Molecular Sciences 25, no. 17: 9636. https://doi.org/10.3390/ijms25179636