

QTL Mapping of Fiber- and Seed-Related Traits in Chromosome Segment Substitution Lines Derived from Gossypium hirsutum × Gossypium darwinii
Abstract
:1. Introduction
2. Results
2.1. Genetic Map Construction
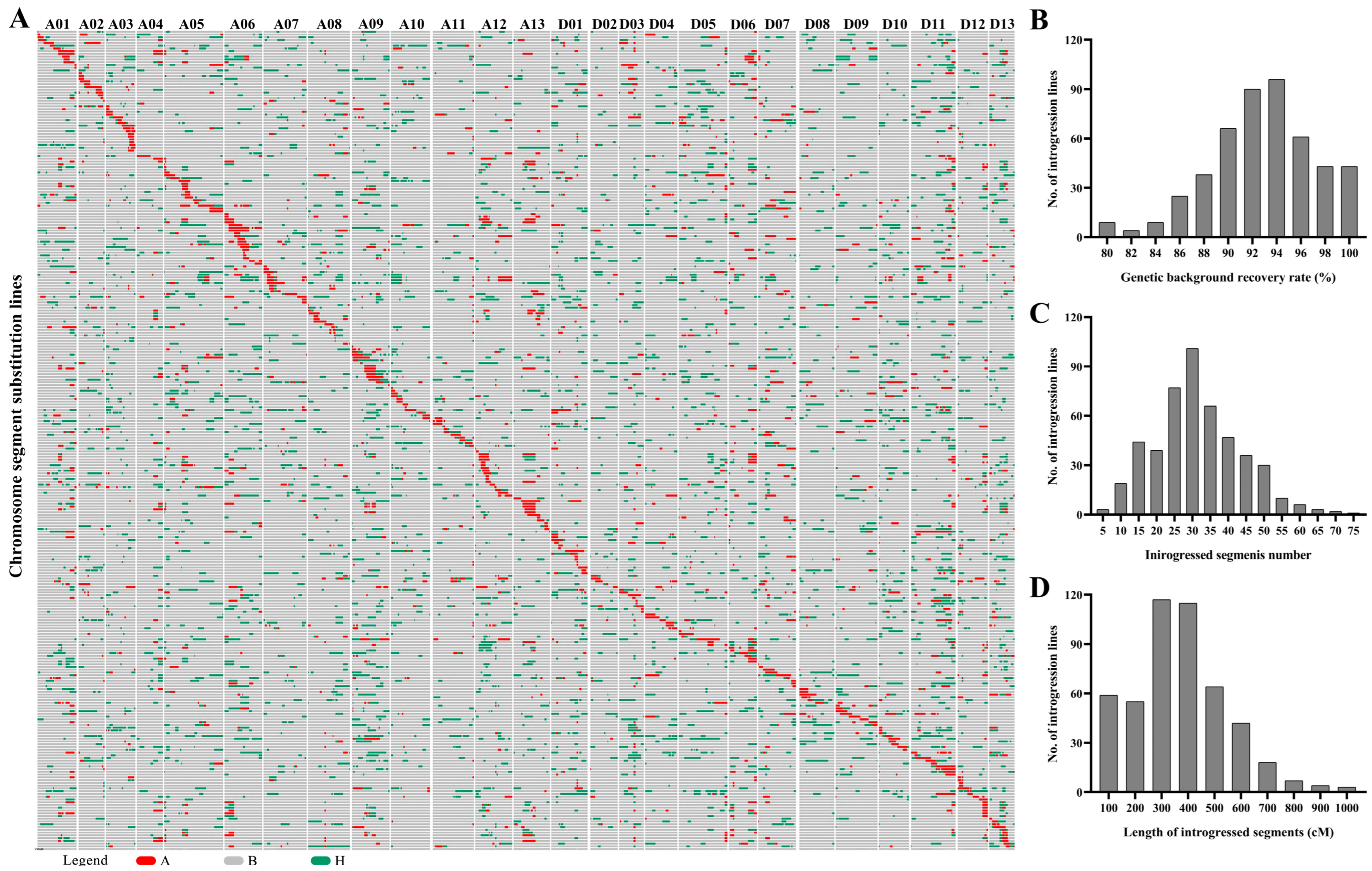
2.2. Introgressive Segments Analysis of the CSSLs
2.3. Characterization of Phenotypic Performance
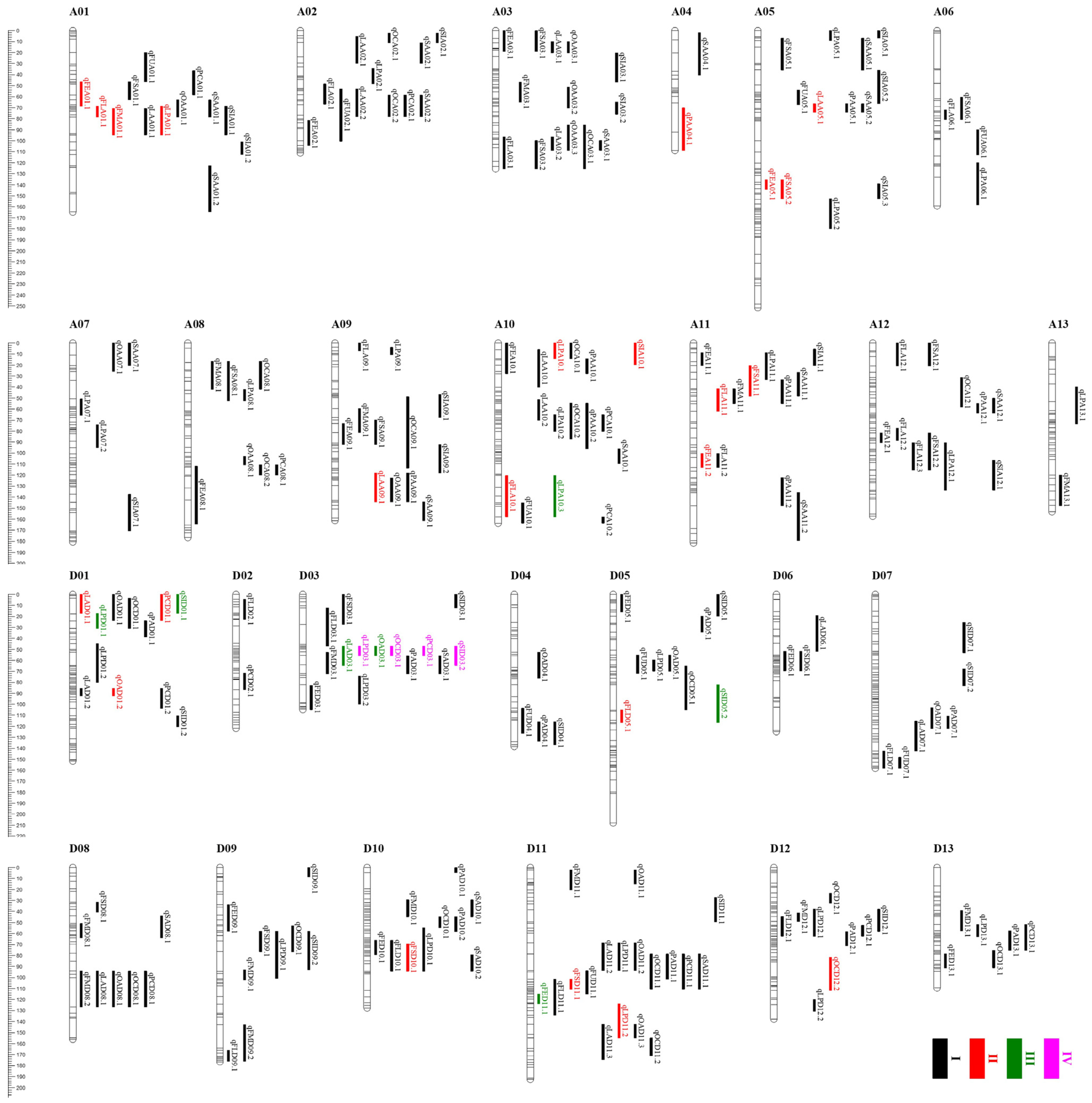
2.4. QTL Mapping for Fiber- and Seed-Related Traits
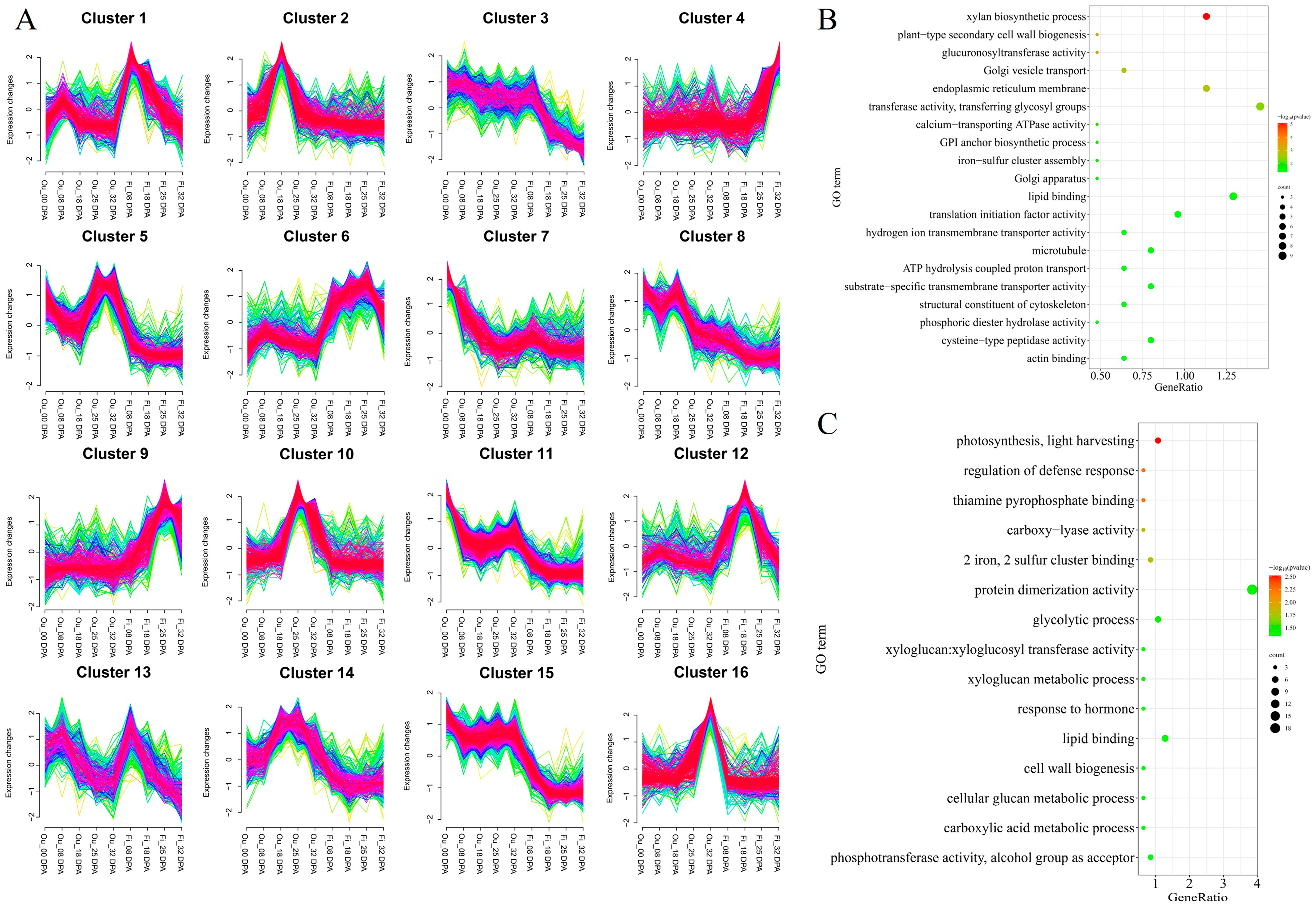
2.5. Functional Annotation of Candidate Genes in QTL Clusters
3. Discussion
3.1. Utilization of G. darwinii and Its Related CSSLs
3.2. Sources and Effects of Favorable Alleles
3.3. Comparison of CSSLs between G. darwinii and G. barbadense
3.4. Identification of Candidate Genes Associated with Stable QTLs
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotypic Collection and Analysis
4.3. Genetic Map Construction and Collinearity Analysis
4.4. Detection of Introgressive Chromosome Segments
4.5. Identification of Fiber- and Seed-Related QTLs
4.6. Functional Annotation of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Khan, N.U.; Basal, H.; Hassan, G. Cottonseed oil and yield assessment via economic heterosis and heritability in intraspecific cotton populations. Afr. J. Biotechnol. 2010, 9, 7418–7428. [Google Scholar]
- Lee, J.H.; Kim, S.H. Synthesis and characterization of biopolyurethane crosslinked with castor oil-based hyperbranched polyols as polymeric solid-solid phase change materials. Sci. Rep. 2022, 12, 14646. [Google Scholar]
- Shen, X.; Guo, W.; Zhu, X.; Yuan, Y.; Yu, J.; Kohel, R.; Zhang, T. Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol. Breed. 2005, 15, 169–181. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Jamshed, M.; Shi, Y.; Liu, A.; Gong, J.; Wang, S.; Zhang, J.; Sun, F.; Jia, F.; et al. Genome-wide quantitative trait loci reveal the genetic basis of cotton fibre quality and yield-related traits in a recombinant inbred line population. Plant Biotechnol. J. 2020, 18, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F.; Grover, C.E. Taxonomy and evolution of the cotton genus, Gossypium. Cotton 2015, 57, 25–44. [Google Scholar]
- Liu, X.; Ma, J.; Li, Q.; Guo, Z.; Wang, Y.; Wang, Q.; Yao, J.; Zhang, Y.; Wang, W.; Teng, Z.; et al. Detection of QTL controlling fiber-related traits in a recombinant inbred lines population from G. hirsutum race punctatum using RTM-GWAS procedure. Ind. Crops Prod. 2023, 193, 116198. [Google Scholar] [CrossRef]
- Wang, B.; Nie, Y.; Lin, Z.; Zhang, X.; Liu, J.; Bai, J. Molecular diversity, genomic constitution, and QTL mapping of fiber quality by mapped SSRs in introgression lines derived from Gossypium hirsutum × G. darwinii Watt. Theor. Appl. Genet. 2012, 125, 1263–1274. [Google Scholar] [CrossRef]
- Grover, C.E.; Zhu, X.; Grupp, K.K.; Jareczek, J.J.; Gallagher, J.P.; Szadkowski, E.; Seijo, J.G.; Wendel, J.F. Molecular confirmation of species status for the allopolyploid cotton species, Gossypium ekmanianum Wittmack. Genet. Resour. Crop Evol. 2015, 62, 103–114. [Google Scholar] [CrossRef]
- Chang, X.; Guo, C.; Pan, Z.; Wu, Y.; Shen, C.; Chao, L.; Shui, G.; You, C.; Xu, J.; Lin, Z.; et al. QTL Mapping for fiber quality based on introgression lines population from G. hirsutum × G. tomentosum. Agriculture 2023, 13, 579. [Google Scholar] [CrossRef]
- Hao, Y.; Liu, X.; Wang, Q.; Wang, S.; Li, Q.; Wang, Y.; Guo, Z.; Wu, T.; Yang, Q.; Bai, Y.; et al. Mapping QTL for fiber-and seed-related traits in Gossypium tomentosum CSSLs with G. hirsutum background. J. Integr. Agric. 2024; in press. [Google Scholar] [CrossRef]
- Wendel, J.F.; Percy, R.G. Allozyme diversity and introgression in the Galapagos Islands endemic Gossypium darwinii and its relationship to continental G. barbadense. Biochem. Syst. Ecol. 1990, 18, 517–528. [Google Scholar] [CrossRef]
- Liang, Z. Genetic and Breeding Science of Cotton Hybridization; Science Press: Beijing, China, 1999. [Google Scholar]
- Jiang, C.X.; Chee, P.W.; Draye, X.; Morrell, P.L.; Paterson, A.H. Multilocus interactions restrict gene introgression in interspecific populations of polyploid Gossypium (cotton). Evolution 2000, 54, 798–814. [Google Scholar]
- Paterson, A.H.; Deverna, J.W.; Lanini, B.; Tanksley, S.D. Fine Mapping of quantitative trait loci using selected overlapping recombinant chromosomes in an interspecies cross of tomato. Genetics 1990, 124, 735–742. [Google Scholar] [CrossRef]
- Young, N.D.; Tanksley, S.D. RFLP analysis of the size of chromosomal segments retained around the Tm-2 locus of tomato during backcross breeding. Theor. Appl. Genet. 1989, 77, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.L.; Sanchez, P.L.; Yu, S.B.; Lorieux, M.; Eizenga, G.C. Chromosome segment substitution lines: A powerful tool for the introgression of valuable genes from Oryza wild species into cultivated rice (O. sativa). Rice 2010, 3, 218–234. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef]
- Song, J.; Pei, W.; Wang, N.; Ma, J.; Xin, Y.; Yang, S.; Wang, W.; Chen, Q.; Zhang, J.; Yu, J.; et al. Transcriptome analysis and identification of genes associated with oil accumulation in upland cotton. Physiol. Plant. 2022, 174, e13701. [Google Scholar] [CrossRef]
- Keerio, A.A.; Shen, C.; Nie, Y.; Ahmed, M.M.; Zhang, X.; Lin, Z. QTL mapping for fiber quality and yield traits based on introgression lines derived from Gossypium hirsutum × G. tomentosum. Int. J. Mol. Sci. 2018, 19, 243. [Google Scholar] [CrossRef]
- Lu, Q.; Li, P.; Yang, R.; Xiao, X.; Li, Z.; Wu, Q.; Gong, J.; Ge, Q.; Liu, A.; Du, S.; et al. QTL mapping and candidate gene prediction for fiber yield and quality traits in a high-generation cotton chromosome substitution line with Gossypium barbadense segments. Mol. Genet. Genom. 2022, 297, 287–301. [Google Scholar] [CrossRef]
- Wang, B.; Draye, X.; Zhuang, Z.; Zhang, Z.; Liu, M.; Lubbers, E.L.; Jones, D.; May, O.L.; Paterson, A.H.; Chee, P.W. QTL analysis of cotton fiber length in advanced backcross populations derived from a cross between Gossypium hirsutum and G. mustelinum. Theor. Appl. Genet. 2017, 130, 1297–1308. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, J.; Chen, Y.; Zhang, C.; Gong, J.; Song, Z.; Zhou, J.; Wang, J.; Zhao, C.; Jiao, M.; et al. Identification of candidate genes for key fibre-related QTLs and derivation of favourable alleles in Gossypium hirsutum recombinant inbred lines with G. barbadense introgressions. Plant Biotechnol. J. 2020, 18, 707–720. [Google Scholar] [CrossRef]
- Yang, P.; Sun, X.; Liu, X.; Wang, W.; Hao, Y.; Chen, L.; Liu, J.; He, H.; Zhang, T.; Bao, W.; et al. Identification of candidate genes for lint percentage and fiber quality through QTL mapping and transcriptome analysis in an allotetraploid interspecific cotton CSSLs population. Front. Plant Sci. 2022, 13, 882051. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Sreedasyam, A.; Ando, A.; Song, Q.; De Santiago LM, D.; Hulse-Kemp, A.M.; Ding, M.; Ye, W.; Kirkbride, R.C.; Jenkins, J.; et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 2020, 52, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, L.; Wang, J.; Wang, Y.; Guo, Z.; Li, Q.; Yang, J.; Wu, Y.; Chen, L.; Teng, Z.; et al. Analyzing quantitative trait loci for fiber quality and yield-related traits from a recombinant inbred line population with Gossypium hirsutum race palmeri as one parent. Front. Plant Sci. 2022, 12, 817748. [Google Scholar] [CrossRef]
- Zhang, M.; Cao, H.; Xi, J.; Zeng, J.; Huang, J.; Li, B.; Song, S.; Zhao, J.; Pei, Y. Auxin directly upregulates GhRAC13 expression to promote the onset of secondary cell wall deposition in cotton fibers. Front. Plant Sci. 2020, 11, 581983. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Sun, M.; Li, W.; Xu, M.; Shao, L.; Liu, Y.; Zhao, G.; Liu, Z.; Xu, Z.; You, J.; et al. Single-cell RNA-seq reveals fate determination control of an individual fibre cell initiation in cotton (Gossypium hirsutum). Plant Biotechnol. J. 2022, 20, 2372–2388. [Google Scholar] [CrossRef] [PubMed]
- Luzarowska, U.; Ruß, A.-K.; Joubès, J.; Batsale, M.; Szymański, J.; PThirumalaikumar, V.P.; Luzarowski, M.; Wu, S.; Zhu, F.; Endres, N.J.; et al. Hello darkness, my old friend: 3-KETOACYL-COENZYME A SYNTHASE 4 is a branch point in the regulation of triacylglycerol synthesis in Arabidopsis thaliana. Plant Cell 2023, 35, 1984–2005. [Google Scholar] [CrossRef]
- Tan, Z.; Fang, X.; Tang, S.; Zhang, J.; Liu, D.; Teng, Z.; Li, L.; Ni, H.; Zheng, F.; Liu, D.; et al. Genetic map and QTL controlling fiber quality traits in upland cotton (Gossypium hirsutum L.). Euphytica 2015, 203, 615–628. [Google Scholar] [CrossRef]
- Wang, W.; Tan, Z.; Xu, Y.; Zhu, A.; Li, Y.; Yao, J.; Tian, R.; Fang, X.; Liu, X.; Tian, Y.; et al. Chromosome structural variation of two cultivated tetraploid cottons and their ancestral diploid species based on a new high-density genetic map. Sci. Rep. 2017, 7, 7640. [Google Scholar]
- Voorrips, R.; Van, D.; Van Den Heuvel, L.; Ooijen, J.; Van, J.W. JoinMap® 4.0: Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma BV: Wageningen, The Netherlands, 2006. [Google Scholar]
- Tang, H.; Krishnakumar, V.; Zeng, X.; Xu, Z.; Taranto, A.; Lomas, J.S.; Zhang, Y.; Huang, Y.; Wang, Y.; Yim, W.C.; et al. JCVI: A versatile toolkit for comparative genomics analysis. iMeta 2024, 3, e211. [Google Scholar] [CrossRef]
- Berloo, R.V. GGT 2.0: Versatile software for visualization and analysis of genetic data. J. Hered. 2008, 99, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, J.W. MapQTL 6.0, Software for the Mapping of Quantitative Trait Loci in Experimental Populations of Dihaploid Species; Kyazma BV: Wageningen, The Netherlands, 2009; Volume 64. [Google Scholar]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17, 101. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | QTLs |
|---|---|
| A01-cluster-1 | qPCA01.1, qFSA01.1, qFEA01.1 |
| A01-cluster-2 | qOAA01.1, qLPA01.1, qFLA01.1, qSIA01.1, qSAA01.1, qFMA01.1, qLAA01.1 |
| A02-cluster-1 | qSIA02.1, qOCA02.1, qSAA02.1, qLAA02.1 |
| A02-cluster-2 | qFLA02.1, qLAA02.2, qSAA02.2, qPCA02.1, qOCA02.2, qFUA02.1 |
| A03-cluster-1 | qFSA03.1, qFEA03.1, qOAA03.1, qLAA03.1 |
| A03-cluster-2 | qFLA03.1, qFSA03.2, qOCA03.1, qSAA03.1, qOAA03.3, qLAA03.2 |
| A05-cluster-1 | qFUA05.1, qPAA05.1, qSAA05.2, qLAA05.1 |
| A08-cluster-1 | qFSA08.1, qFMA08.1, qOCA08.1, qLPA08.1 |
| A09-cluster-1 | qPAA09.1, qOAA09.1, qLAA09.1 |
| A10-cluster-1 | qLPA10.1, qSIA10.1, qOCA10.1, qFEA10.1, qPAA10.1 |
| A10-cluster-2 | qPAA10.2, qLAA10.2, qPCA10.1, qOCA10.2, qLPA10.2 |
| A11-cluster-1 | qSIA11.1, qFSA11.1, qFEA11.1, qLPA11.1 |
| A11-cluster-2 | qFLA11.1, qFMA11.1, qPAA11.1 |
| A12-cluster-1 | qOCA12.1, qPAA12.1, qSAA12.1 |
| A12-cluster-2 | qFLA12.3, qFSA12.2, qLPA12.1, qSIA12.1 |
| D01-cluster-1 | qPCD01.1, qSID01.1, qOAD01.1, qLAD01.1, qOCD01.1 |
| D01-cluster-2 | qOAD01.2, qPCD01.2, qLAD01.2 |
| D03-cluster-1 | qLPD03.1, qPCD03.1, qOCD03.1, qOAD03.1, qLAD03.1, qSID03.2, qFMD03.1, qPAD03.1, qSAD03.1 |
| D05-cluster-1 | qLPD05.1, qFUD05.1, qOAD05.1 |
| D05-cluster-2 | qFLD05.1, qFSD05.1, qSID05.2 |
| D08-cluster-1 | qFMD08.2, qPCD08.1, qOCD08.1, qOAD08.1, qLAD08.1 |
| D09-cluster-1 | qSID09.2, qFSD09.1, qOCD09.1, qLPD09.1 |
| D10-cluster-1 | qLPD10.1, qFED10.1, qFLD10.1, qFSD10.1, qSAD10.2 |
| D11-cluster-1 | qLPD11.1, qOAD11.2, qLAD11.2, qPCD11.1, qOCD11.1, qPAD11.1, qSAD11.1 |
| D12-cluster-1 | qLPD12.1, qFLD12.1, qSID12.1, qPCD12.1, qPAD12.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Li, Y.; Le, M.; Tian, L.; Sun, X.; Liu, R.; Guo, X.; Wu, Y.; Li, Y.; Zhao, J.; et al. QTL Mapping of Fiber- and Seed-Related Traits in Chromosome Segment Substitution Lines Derived from Gossypium hirsutum × Gossypium darwinii. Int. J. Mol. Sci. 2024, 25, 9639. https://doi.org/10.3390/ijms25179639
Wang W, Li Y, Le M, Tian L, Sun X, Liu R, Guo X, Wu Y, Li Y, Zhao J, et al. QTL Mapping of Fiber- and Seed-Related Traits in Chromosome Segment Substitution Lines Derived from Gossypium hirsutum × Gossypium darwinii. International Journal of Molecular Sciences. 2024; 25(17):9639. https://doi.org/10.3390/ijms25179639
Chicago/Turabian StyleWang, Wenwen, Yan Li, Mingmei Le, Lixia Tian, Xujing Sun, Rui Liu, Xin Guo, Yan Wu, Yibing Li, Jiaoyun Zhao, and et al. 2024. "QTL Mapping of Fiber- and Seed-Related Traits in Chromosome Segment Substitution Lines Derived from Gossypium hirsutum × Gossypium darwinii" International Journal of Molecular Sciences 25, no. 17: 9639. https://doi.org/10.3390/ijms25179639