
Metabolic Adaptation in Epilepsy: From Acute Response to Chronic Impairment
 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Acute Adaptation of Neuronal Slices to Prolonged Seizure-like Activity
2.2. Metabolic Effects
2.3. Metabolic Characterization of Epileptic Brain Tissue
2.3.1. Maximal ATP Production Capacities
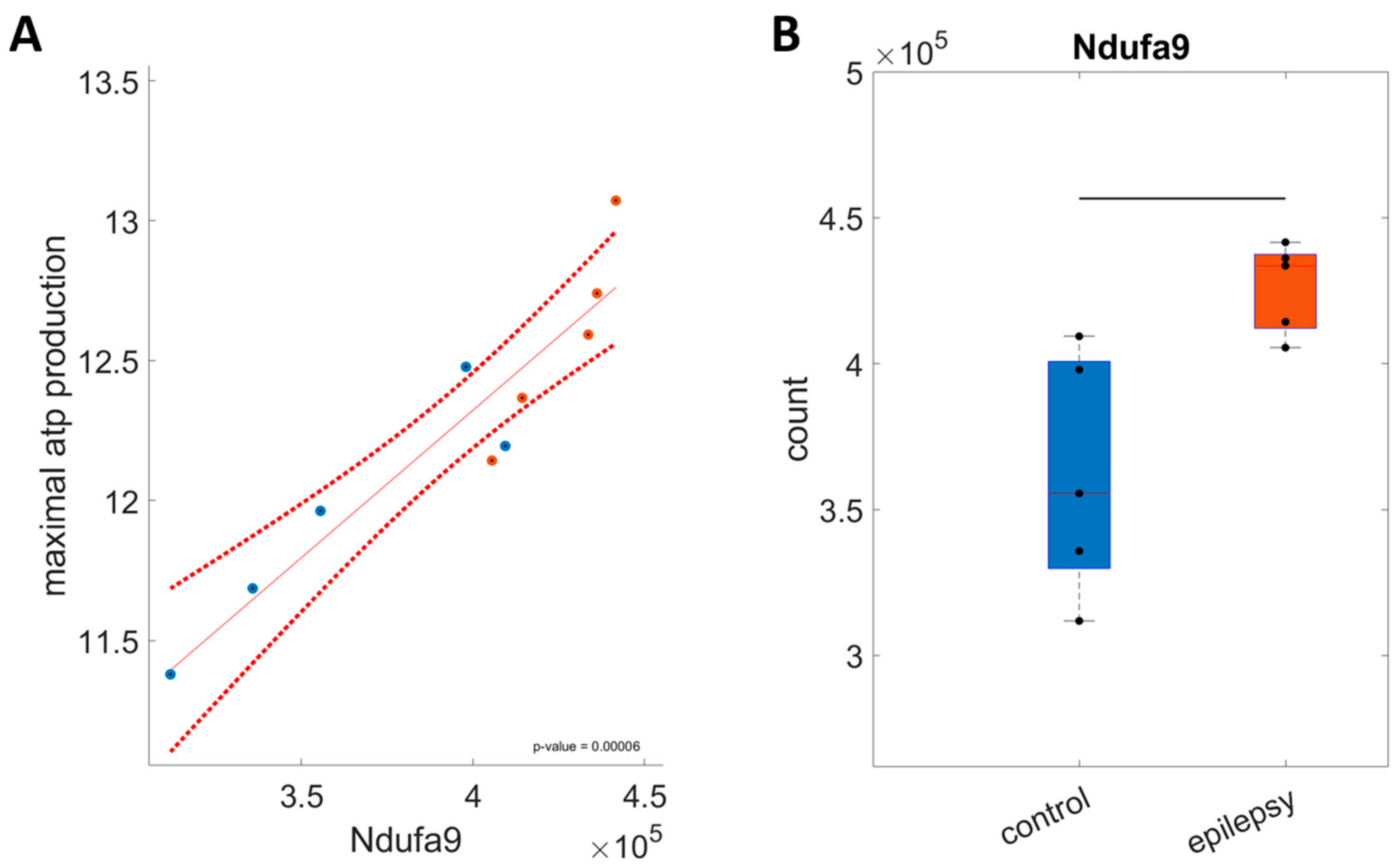
2.3.2. Coordinated Upregulation of Respiratory Chain and Oxidative Phosphorylation Enzymes
2.3.3. Metabolic Changes in Chronic Epileptic Tissues
3. Discussion
4. Materials and Methods
4.1. Slice Preparation, Maintenance, and Induction of Seizure Activity
4.2. Electrophysiology and ptiO2 Recordings
4.3. Data Acquisition and Data Analysis
4.4. Calculation of Cerebral Metabolic Rate of O2
4.5. Proteomics Sample Preparation
4.6. Liquid Chromatography–Mass Spectrometry (LC–MS)
4.7. Generation of the Spectral Library
4.8. Data Analysis
4.9. Metabolic Modeling
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ames, A., 3rd. CNS energy metabolism as related to function. Brain Res. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef] [PubMed]
- Schoknecht, K.; Berndt, N.; Rosner, J.; Heinemann, U.; Dreier, J.P.; Kovacs, R.; Friedman, A.; Liotta, A. Event-Associated Oxygen Consumption Rate Increases ca. Five-Fold When Interictal Activity Transforms into Seizure-Like Events In Vitro. Int. J. Mol. Sci. 2017, 18, 1925. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I.; Bernard, C.; Turner, D.A. Metabolic responses differentiate between interictal, ictal and persistent epileptiform activity in intact, immature hippocampus in vitro. Neurobiol. Dis. 2015, 75, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Alavi, A.; Yakir, S.; Newberg, A.B. Positron emission tomography in seizure disorders. Ann. N. Y. Acad. Sci. 2011, 1228, E1–E12. [Google Scholar] [CrossRef]
- Pan, J.W.; Williamson, A.; Cavus, I.; Hetherington, H.P.; Zaveri, H.; Petroff, O.A.; Spencer, D.D. Neurometabolism in human epilepsy. Epilepsia 2008, 49 (Suppl. S3), 31–41. [Google Scholar] [CrossRef]
- Vielhaber, S.; Von Oertzen, J.H.; Kudin, A.F.; Schoenfeld, A.; Menzel, C.; Biersack, H.J.; Kral, T.; Elger, C.E.; Kunz, W.S. Correlation of hippocampal glucose oxidation capacity and interictal FDG-PET in temporal lobe epilepsy. Epilepsia 2003, 44, 193–199. [Google Scholar] [CrossRef]
- Kovacs, R.; Heinemann, U.; Steinhauser, C. Mechanisms underlying blood-brain barrier dysfunction in brain pathology and epileptogenesis: Role of astroglia. Epilepsia 2012, 53 (Suppl. S6), 53–59. [Google Scholar] [CrossRef]
- Angamo, E.A.; ul Haq, R.; Rosner, J.; Gabriel, S.; Gerevich, Z.; Heinemann, U.; Kovacs, R. Contribution of Intrinsic Lactate to Maintenance of Seizure Activity in Neocortical Slices from Patients with Temporal Lobe Epilepsy and in Rat Entorhinal Cortex. Int. J. Mol. Sci. 2017, 18, 1835. [Google Scholar] [CrossRef]
- Bedner, P.; Dupper, A.; Huttmann, K.; Muller, J.; Herde, M.K.; Dublin, P.; Deshpande, T.; Schramm, J.; Haussler, U.; Haas, C.A.; et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 2015, 138 Pt 5, 1208–1222. [Google Scholar] [CrossRef]
- Lauritzen, F.; Heuser, K.; de Lanerolle, N.C.; Lee, T.S.; Spencer, D.D.; Kim, J.H.; Gjedde, A.; Eid, T.; Bergersen, L.H. Redistribution of monocarboxylate transporter 2 on the surface of astrocytes in the human epileptogenic hippocampus. Glia 2012, 60, 1172–1181. [Google Scholar] [CrossRef]
- Berndt, N.; Kann, O.; Holzhutter, H.G. Physiology-based kinetic modeling of neuronal energy metabolism unravels the molecular basis of NAD(P)H fluorescence transients. J. Cereb. Blood Flow Metab. 2015, 35, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Canto, A.M.; Matos, A.H.B.; Godoi, A.B.; Vieira, A.S.; Aoyama, B.B.; Rocha, C.S.; Henning, B.; Carvalho, B.S.; Pascoal, V.D.B.; Veiga, D.F.T.; et al. Multi-omics analysis suggests enhanced epileptogenesis in the Cornu Ammonis 3 of the pilocarpine model of mesial temporal lobe epilepsy. Hippocampus 2021, 31, 122–139. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Jia, Y.; Zhao, Y.; Ma, C.; Bao, X.; Meng, X.; Dou, W.; Wang, X.; Ge, W. Proteomic profiling of sclerotic hippocampus revealed dysregulated packaging of vesicular neurotransmitters in temporal lobe epilepsy. Epilepsy Res. 2020, 166, 106412. [Google Scholar] [CrossRef]
- Kjaer, C.; Barzaghi, G.; Bak, L.K.; Goetze, J.P.; Yde, C.W.; Woldbye, D.; Pinborg, L.H.; Jensen, L.J. Transcriptome analysis in patients with temporal lobe epilepsy. Brain 2019, 142, e55. [Google Scholar] [CrossRef] [PubMed]
- Antonio, L.L.; Anderson, M.L.; Angamo, E.A.; Gabriel, S.; Klaft, Z.J.; Liotta, A.; Salar, S.; Sandow, N.; Heinemann, U. In vitro seizure like events and changes in ionic concentration. J. Neurosci. Methods 2016, 260, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Holzhutter, H.G. Dynamic Metabolic Zonation of the Hepatic Glucose Metabolism Is Accomplished by Sinusoidal Plasma Gradients of Nutrients and Hormones. Front. Physiol. 2018, 9, 1786. [Google Scholar] [CrossRef]
- Bulik, S.; Holzhutter, H.G.; Berndt, N. The relative importance of kinetic mechanisms and variable enzyme abundances for the regulation of hepatic glucose metabolism--insights from mathematical modeling. BMC Biol. 2016, 14, 15. [Google Scholar] [CrossRef]
- Browning, J.D.; Baxter, J.; Satapati, S.; Burgess, S.C. The effect of short-term fasting on liver and skeletal muscle lipid, glucose, and energy metabolism in healthy women and men. J. Lipid Res. 2012, 53, 577–586. [Google Scholar] [CrossRef]
- Shi, X.; Qiu, H. New Insights Into Energy Substrate Utilization and Metabolic Remodeling in Cardiac Physiological Adaption. Front. Physiol. 2022, 13, 831829. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Hudert, C.A.; Eckstein, J.; Loddenkemper, C.; Henning, S.; Bufler, P.; Meierhofer, D.; Sack, I.; Wiegand, S.; Wallach, I.; et al. Alterations of Central Liver Metabolism of Pediatric Patients with Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 11072. [Google Scholar] [CrossRef]
- Berndt, N.; Eckstein, J.; Wallach, I.; Nordmeyer, S.; Kelm, M.; Kirchner, M.; Goubergrits, L.; Schafstedde, M.; Hennemuth, A.; Kraus, M.; et al. CARDIOKIN1: Computational Assessment of Myocardial Metabolic Capability in Healthy Controls and Patients With Valve Diseases. Circulation 2021, 144, 1926–1939. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Chan, P. Skeletal Muscle Metabolic Alternation Develops Sarcopenia. Aging Dis. 2022, 13, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Puchowicz, M.A.; Koppaka, S.S.; LaManna, J.C. Brain Metabolic Adaptations to Hypoxia. In Metabolic Encephalopathy; McCandless, D., Ed.; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Beland-Millar, A.; Takimoto, M.; Hamada, T.; Messier, C. Brain and muscle adaptation to high-fat diets and exercise: Metabolic transporters, enzymes and substrates in the rat cortex and muscle. Brain Res. 2020, 1749, 147126. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, R.; Schuchmann, S.; Gabriel, S.; Kann, O.; Kardos, J.; Heinemann, U. Free radical-mediated cell damage after experimental status epilepticus in hippocampal slice cultures. J. Neurophysiol. 2002, 88, 2909–2918. [Google Scholar] [CrossRef]
- Kann, O.; Kovacs, R.; Njunting, M.; Behrens, C.J.; Otahal, J.; Lehmann, T.N.; Gabriel, S.; Heinemann, U. Metabolic dysfunction during neuronal activation in the ex vivo hippocampus from chronic epileptic rats and humans. Brain 2005, 128 Pt 10, 2396–2407. [Google Scholar] [CrossRef]
- Malinska, D.; Kulawiak, B.; Kudin, A.P.; Kovacs, R.; Huchzermeyer, C.; Kann, O.; Szewczyk, A.; Kunz, W.S. Complex III-dependent superoxide production of brain mitochondria contributes to seizure-related ROS formation. Biochim. Biophys. Acta 2010, 1797, 1163–1170. [Google Scholar] [CrossRef]
- Rowley, S.; Patel, M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Radic. Biol. Med. 2013, 62, 121–131. [Google Scholar] [CrossRef]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Kunz, W.S.; Kudin, A.P.; Vielhaber, S.; Blumcke, I.; Zuschratter, W.; Schramm, J.; Beck, H.; Elger, C.E. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 2000, 48, 766–773. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Lublinsky, S.; Kamintsky, L.; Noyman, I.; Veksler, R.; Dalipaj, H.; Senatorov, V.V., Jr.; Swissa, E.; Rosenbach, D.; Elazary, N.; et al. Imaging blood-brain barrier dysfunction as a biomarker for epileptogenesis. Brain 2017, 140, 1692–1705. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, U.; Kaufer, D.; Friedman, A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia 2012, 60, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Weissberg, I.; Wood, L.; Kamintsky, L.; Vazquez, O.; Milikovsky, D.Z.; Alexander, A.; Oppenheim, H.; Ardizzone, C.; Becker, A.; Frigerio, F.; et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-beta/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol. Dis. 2015, 78, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Prager, O.; Kamintsky, L.; Hasam-Henderson, L.A.; Schoknecht, K.; Wuntke, V.; Papageorgiou, I.; Swolinsky, J.; Muoio, V.; Bar-Klein, G.; Vazana, U.; et al. Seizure-induced microvascular injury is associated with impaired neurovascular coupling and blood-brain barrier dysfunction. Epilepsia 2019, 60, 322–336. [Google Scholar] [CrossRef]
- Parfenova, H.; Carratu, P.; Tcheranova, D.; Fedinec, A.; Pourcyrous, M.; Leffler, C.W. Epileptic seizures cause extended postictal cerebral vascular dysfunction that is prevented by HO-1 overexpression. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2843–H2850. [Google Scholar] [CrossRef]
- Milesi, S.; Boussadia, B.; Plaud, C.; Catteau, M.; Rousset, M.C.; De Bock, F.; Schaeffer, M.; Lerner-Natoli, M.; Rigau, V.; Marchi, N. Redistribution of PDGFRbeta cells and NG2DsRed pericytes at the cerebrovasculature after status epilepticus. Neurobiol. Dis. 2014, 71, 151–158. [Google Scholar] [CrossRef]
- Leal-Campanario, R.; Alarcon-Martinez, L.; Rieiro, H.; Martinez-Conde, S.; Alarcon-Martinez, T.; Zhao, X.; LaMee, J.; Popp, P.J.; Calhoun, M.E.; Arribas, J.I.; et al. Abnormal Capillary Vasodynamics Contribute to Ictal Neurodegeneration in Epilepsy. Sci. Rep. 2017, 7, 43276. [Google Scholar] [CrossRef]
- Farrell, J.S.; Colangeli, R.; Wolff, M.D.; Wall, A.K.; Phillips, T.J.; George, A.; Federico, P.; Teskey, G.C. Postictal hypoperfusion/hypoxia provides the foundation for a unified theory of seizure-induced brain abnormalities and behavioral dysfunction. Epilepsia 2017, 58, 1493–1501. [Google Scholar] [CrossRef]
- Berndt, N.; Rosner, J.; Haq, R.U.; Kann, O.; Kovacs, R.; Holzhutter, H.G.; Spies, C.; Liotta, A. Possible neurotoxicity of the anesthetic propofol: Evidence for the inhibition of complex II of the respiratory chain in area CA3 of rat hippocampal slices. Arch. Toxicol. 2018, 92, 3191–3205. [Google Scholar] [CrossRef]
- Guillon, B.; Duncan, R.; Biraben, A.; Bernard, A.M.; Vignal, J.P.; Chauvel, P. Correlation between interictal regional cerebral blood flow and depth-recorded interictal spiking in temporal lobe epilepsy. Epilepsia 1998, 39, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Suh, M.; Bahar, S.; Mehta, A.D.; Schwartz, T.H. Temporal dependence in uncoupling of blood volume and oxygenation during interictal epileptiform events in rat neocortex. J. Neurosci. 2005, 25, 68–77. [Google Scholar] [CrossRef]
- Geneslaw, A.S.; Zhao, M.; Ma, H.; Schwartz, T.H. Tissue hypoxia correlates with intensity of interictal spikes. J. Cereb. Blood Flow Metab. 2011, 31, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Reiffurth, C.; Berndt, N.; Gonzalez Lopez, A.; Schoknecht, K.; Kovacs, R.; Maechler, M.; Grote Lambers, M.; Dreier, J.P.; Friedman, A.; Spies, C.; et al. Deep Isoflurane Anesthesia Is Associated with Alterations in Ion Homeostasis and Specific Na+/K+-ATPase Impairment in the Rat Brain. Anesthesiology 2023, 138, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. Cell death and synaptic reorganizations produced by seizures. Epilepsia 2001, 42 (Suppl. S3), 5–7. [Google Scholar] [CrossRef]
- Jayakumar, S.; Hasan, G. Neuronal Calcium Signaling in Metabolic Regulation and Adaptation to Nutrient Stress. Front. Neural Circuits 2018, 12, 25. [Google Scholar] [CrossRef]
- Naquet, P.; Giessner, C.; Galland, F. Metabolic adaptation of tissues to stress releases metabolites influencing innate immunity. Curr. Opin. Immunol. 2016, 38, 30–38. [Google Scholar] [CrossRef]
- Vettor, R.; Di Vincenzo, A.; Maffei, P.; Rossato, M. Regulation of energy intake and mechanisms of metabolic adaptation or maladaptation after caloric restriction. Rev. Endocr. Metab. Disord. 2020, 21, 399–409. [Google Scholar] [CrossRef]
- do Nascimento, A.L.; Dos Santos, N.F.; Campos Pelagio, F.; Aparecida Teixeira, S.; de Moraes Ferrari, E.A.; Langone, F. Neuronal degeneration and gliosis time-course in the mouse hippocampal formation after pilocarpine-induced status epilepticus. Brain Res. 2012, 1470, 98–110. [Google Scholar] [CrossRef]
- Henshall, D.C.; Engel, T. Contribution of apoptosis-associated signaling pathways to epileptogenesis: Lessons from Bcl-2 family knockouts. Front. Cell. Neurosci. 2013, 7, 110. [Google Scholar] [CrossRef]
- Liotta, A.; Rosner, J.; Huchzermeyer, C.; Wojtowicz, A.; Kann, O.; Schmitz, D.; Heinemann, U.; Kovacs, R. Energy demand of synaptic transmission at the hippocampal Schaffer-collateral synapse. J. Cereb. Blood Flow Metab. 2012, 32, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Huchzermeyer, C.; Berndt, N.; Holzhutter, H.G.; Kann, O. Oxygen consumption rates during three different neuronal activity states in the hippocampal CA3 network. J. Cereb. Blood Flow Metab. 2013, 33, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Kasischke, K.A.; Lambert, E.M.; Panepento, B.; Sun, A.; Gelbard, H.A.; Burgess, R.W.; Foster, T.H.; Nedergaard, M. Two-photon NADH imaging exposes boundaries of oxygen diffusion in cortical vascular supply regions. J. Cereb. Blood Flow Metab. 2011, 31, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liotta, A.; Loroch, S.; Wallach, I.; Klewe, K.; Marcus, K.; Berndt, N. Metabolic Adaptation in Epilepsy: From Acute Response to Chronic Impairment. Int. J. Mol. Sci. 2024, 25, 9640. https://doi.org/10.3390/ijms25179640
Liotta A, Loroch S, Wallach I, Klewe K, Marcus K, Berndt N. Metabolic Adaptation in Epilepsy: From Acute Response to Chronic Impairment. International Journal of Molecular Sciences. 2024; 25(17):9640. https://doi.org/10.3390/ijms25179640
Chicago/Turabian StyleLiotta, Agustin, Stefan Loroch, Iwona Wallach, Kristoffer Klewe, Katrin Marcus, and Nikolaus Berndt. 2024. "Metabolic Adaptation in Epilepsy: From Acute Response to Chronic Impairment" International Journal of Molecular Sciences 25, no. 17: 9640. https://doi.org/10.3390/ijms25179640
APA StyleLiotta, A., Loroch, S., Wallach, I., Klewe, K., Marcus, K., & Berndt, N. (2024). Metabolic Adaptation in Epilepsy: From Acute Response to Chronic Impairment. International Journal of Molecular Sciences, 25(17), 9640. https://doi.org/10.3390/ijms25179640