
C-C Chemokine Receptor 7 Promotes T-Cell Acute Lymphoblastic Leukemia Invasion of the Central Nervous System via β2-Integrins

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
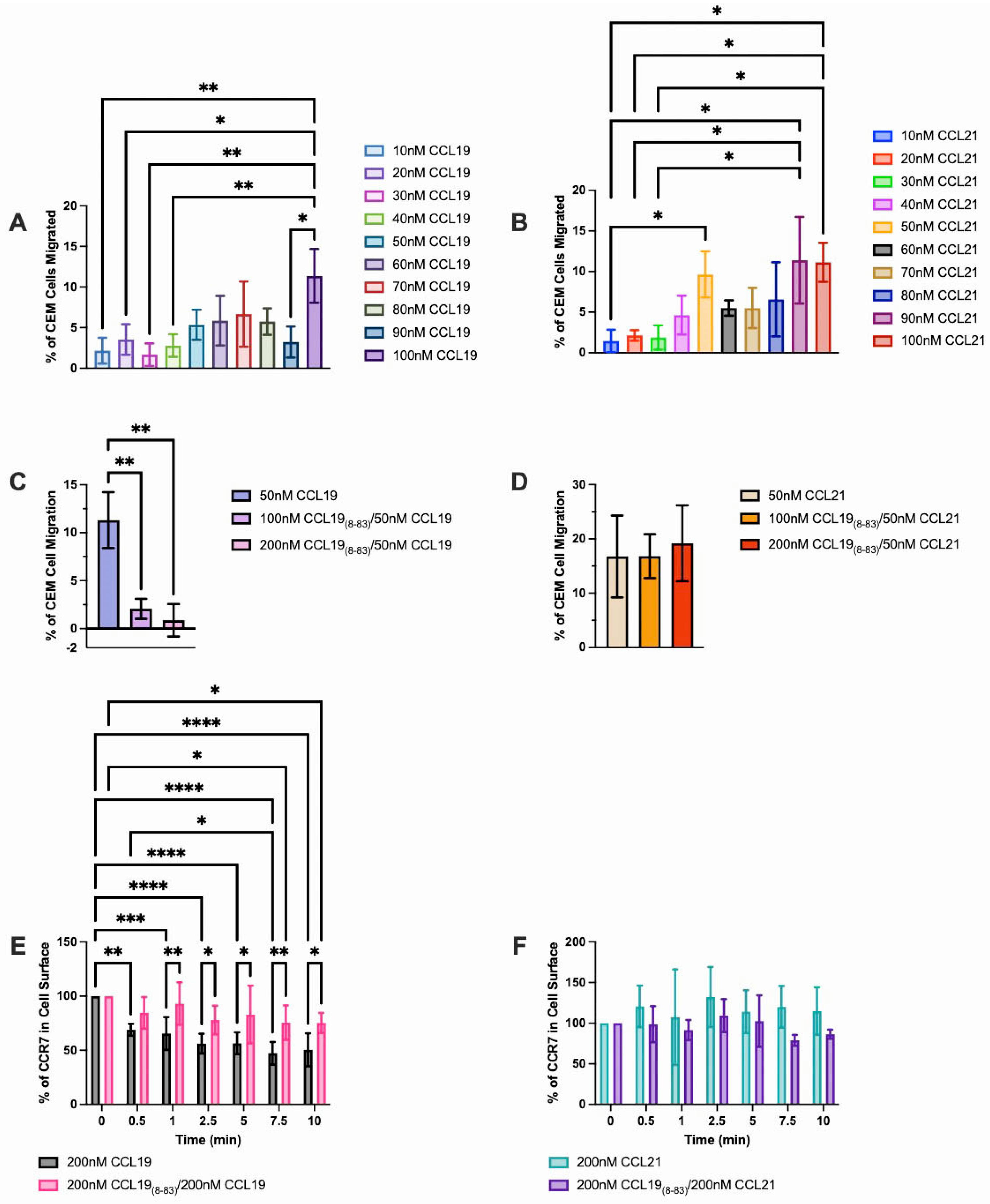
2. Results
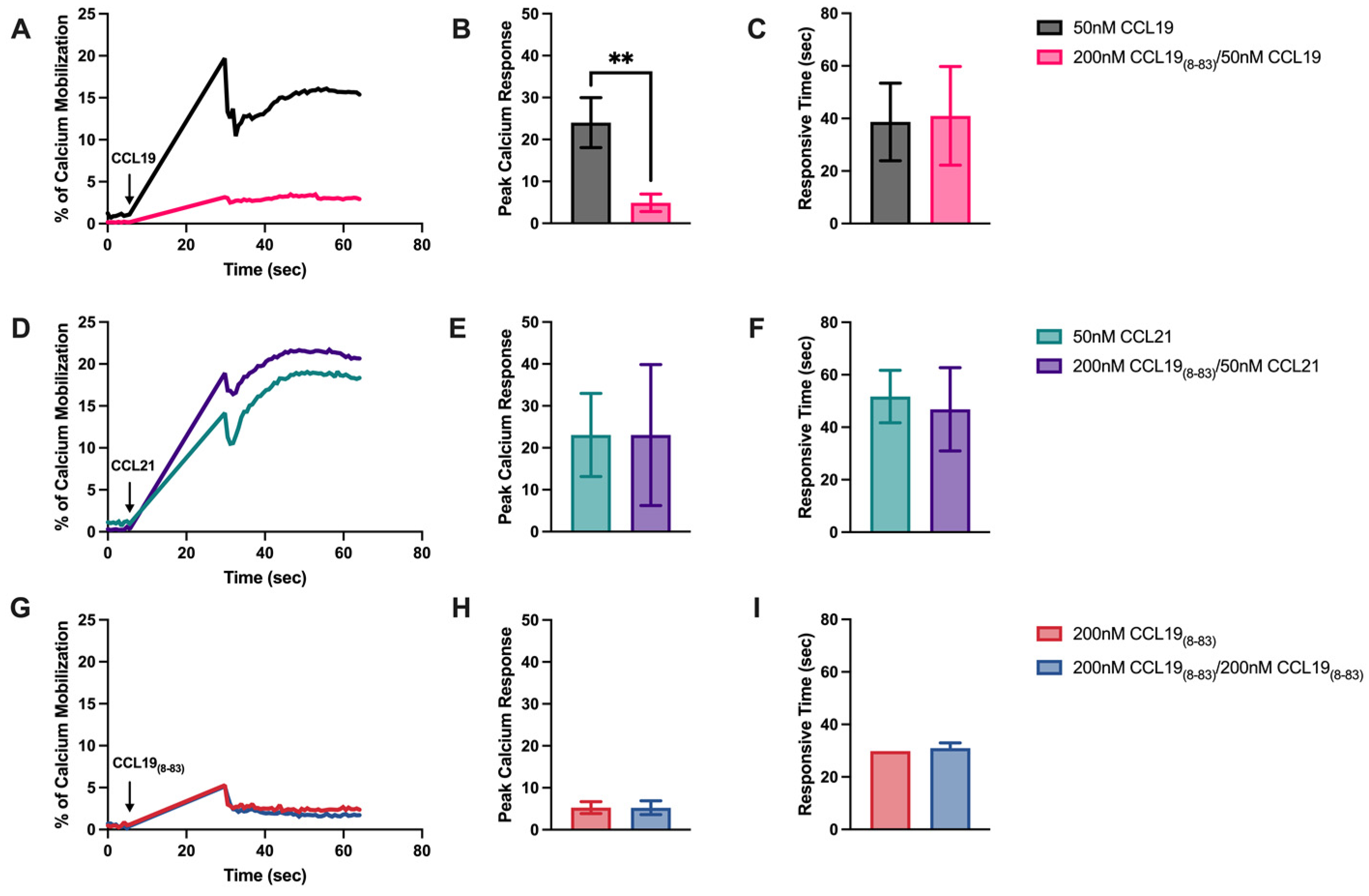
2.1. CCL198-83 Inhibits CCL19-Induced CCR7 Internalization and Signaling but Not CCL21
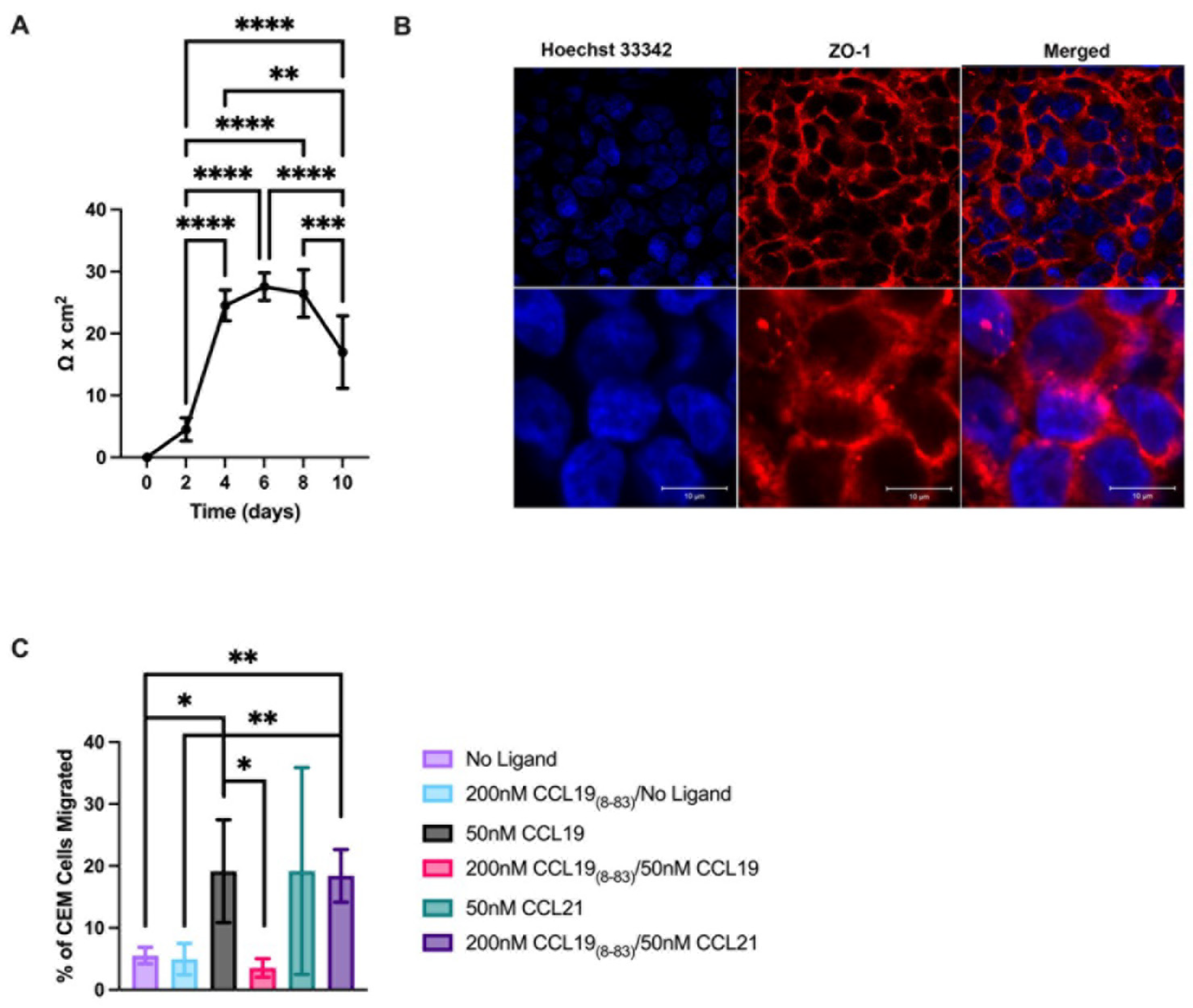
2.2. CCL18-83 Prevents Migration of T-ALL across a Human Endothelial Cell Monolayer
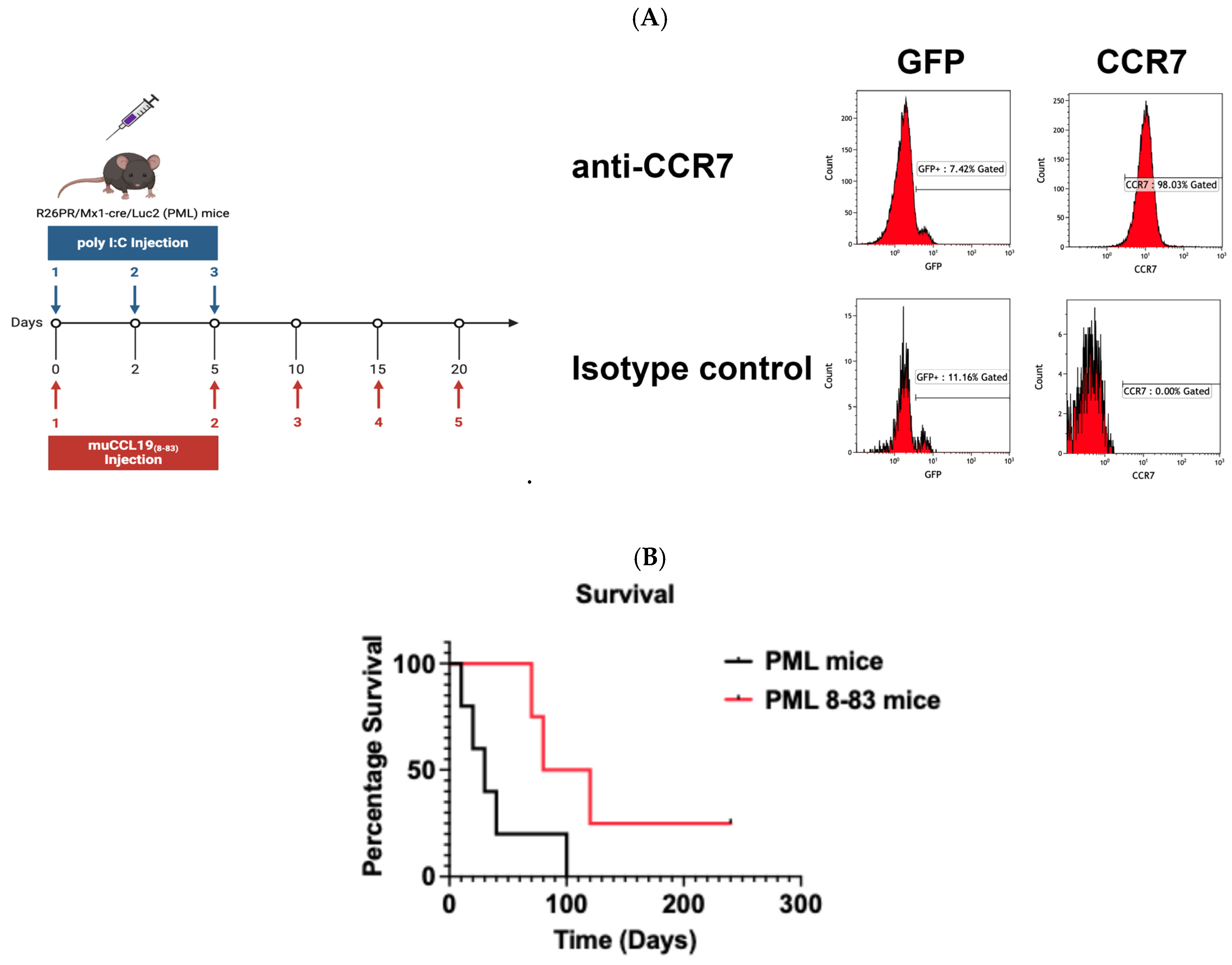
CCL198-83 Prophylactic Treatment Substantially Extends Survival in a Mouse Model of T-ALL
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Cancer Society. Leukemia at a Glance. 2024. Available online: https://cancerstatisticscenter.cancer.org/#!/cancer-site/Leukemia (accessed on 24 July 2024).
- Patel, A.A.; Thomas, J.; Rojek, A.E.; Stock, W. Biology and Treatment Paradigms in T Cell Acute Lymphoblastic Leukemia in Older Adolescents and Adults. Curr. Treat. Options Oncol. 2020, 21, 57. [Google Scholar] [CrossRef]
- Bill, C.A.; Allen, C.M.; Vines, C.M. C-C Chemokine Receptor 7 in Cancer. Cells 2022, 11, 656. [Google Scholar] [CrossRef]
- Ma, W.; Gutierrez, A.; Goff, D.J.; Geron, I.; Sadarangani, A.; Jamieson, C.A.M.; Court, A.C.; Shih, A.Y.; Jiang, Q.; Wu, C.C.; et al. NOTCH1 signaling promotes human T-cell acute lymphoblastic leukemia initiating cell regeneration in supportive niches. PLoS ONE 2012, 7, e39725. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Fijalkowski, I.; Wang, J.; Jin, Q.; Van Laere, J.; Serafin, V.; Crispino, J.D.; Ntziachristos, P. Correction: A positive feedback loop regulation between NOTCH1 and USP11 in T-cell leukemia. Leukemia 2024, 38, 229–232. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Trimarchi, T.; Ruocco, M.G.; Reavie, L.; Cathelin, S.; Mar, B.G.; Klinakis, A.; Lukyanov, Y.; Tseng, J.-C.; Sen, F.; et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature 2009, 459, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Hedrick, J.; Zlotnik, A.; Siani, M.A.; Thompson, D.A.; Butcher, E.C. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science 1998, 279, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Bowman, E.P.; Murphy, K.; Youngman, K.R.; Siani, M.A.; Thompson, D.A.; Wu, L.; Zlotnik, A.; Butcher, E.C. 6-C-kine (SLC), a lymphocyte adhesion-triggering chemokine expressed by high endothelium, is an agonist for the MIP-3beta receptor CCR7. J. Cell Biol. 1998, 141, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Vigh, J.P.; Kincses, A.; Ozgür, B.; Walter, F.R.; Santa-Maria, A.R.; Valkai, S.; Vastag, M.; Neuhaus, W.; Brodin, B.; Dér, A.; et al. Transendothelial Electrical Resistance Measurement across the Blood-Brain Barrier: A Critical Review of Methods. Micromachines 2021, 12, 685. [Google Scholar] [CrossRef]
- Watson, P.M.; Anderson, J.M.; Vanltallie, C.M.; Doctrow, S.R. The tight-junction-specific protein ZO-1 is a component of the human and rat blood-brain barriers. Neurosci. Lett. 1991, 129, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E. Endothelial cell-cell junctions: Happy together. Nat. Rev. Mol. Cell Biol. 2004, 5, 261–270. [Google Scholar] [CrossRef]
- Pieters, R.; Mullighan, C.G.; Hunger, S.P. Advancing Diagnostics and Therapy to Reach Universal Cure in Childhood ALL. J. Clin. Oncol. 2023, 41, 5579–5591. [Google Scholar] [CrossRef] [PubMed]
- Pilkington, K.R.; Clark-Lewis, I.; McColl, S.R. Inhibition of generation of cytotoxic T lymphocyte activity by a CCL19/macrophage inflammatory protein (MIP)-3beta antagonist. J. Biol. Chem. 2004, 279, 40276–40282. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, E.M.; Kroeck, K.G.; Jacobs, L.M.; Fenske, T.G.; Witt, R.N.; Hintz, A.M.; Ramsden, E.R.; Zhang, X.; Peterson, F.; Volkman, B.F.; et al. Structural Insights into Molecular Recognition by Human Chemokine CCL19. Biochemistry 2022, 61, 311–318. [Google Scholar] [CrossRef]
- Smith, E.W.; Lewandowski, E.M.; Moussouras, N.A.; Kroeck, K.G.; Volkman, B.F.; Veldkamp, C.T.; Chen, Y. Crystallographic Structure of Truncated CCL21 and the Putative Sulfotyrosine-Binding Site. Biochemistry 2016, 55, 5746–5753. [Google Scholar] [CrossRef]
- Veldkamp, C.T.; Kiermaier, E.; Gabel-Eissens, S.J.; Gillitzer, M.L.; Lippner, D.R.; DiSilvio, F.A.; Mueller, C.J.; Wantuch, P.L.; Chaffee, G.R.; Famiglietti, M.W.; et al. Solution Structure of CCL19 and Identification of Overlapping CCR7 and PSGL-1 Binding Sites. Biochemistry 2015, 54, 4163–4166. [Google Scholar] [CrossRef]
- Huang, C.-C.; Lam, S.N.; Acharya, P.; Tang, M.; Xiang, S.-H.; Hussan, S.S.-U.; Stanfield, R.L.; Robinson, J.; Sodroski, J.; Wilson, I.A.; et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 2007, 317, 1930–1934. [Google Scholar] [CrossRef]
- Millard, C.J.; Ludeman, J.P.; Canals, M.; Bridgford, J.L.; Hinds, M.G.; Clayton, D.J.; Christopoulos, A.; Payne, R.J.; Stone, M.J. Structural basis of receptor sulfotyrosine recognition by a CC chemokine: The N-terminal region of CCR3 bound to CCL11/eotaxin-1. Structure 2014, 22, 1571–1581. [Google Scholar] [CrossRef]
- Carofino, B.L.; Ayanga, B.; Justice, M.J. A mouse model for inducible overexpression of Prdm14 results in rapid-onset and highly penetrant T-cell acute lymphoblastic leukemia (T-ALL). Dis. Model. Mech. 2013, 6, 1494–1506. [Google Scholar] [CrossRef]
- Carofino, B.L.; Ayanga, B.; Tracey, L.J.; Brooke-Bisschop, T.; Justice, M.J. PRDM14 promotes RAG-dependent Notch1 driver mutations in mouse T-ALL. Biol. Open 2016, 5, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Somersalo, K.; Tarkkanen, J.; Patarroyo, M.; Saksela, E. Involvement of beta 2-integrins in the migration of human natural killer cells. J. Immunol. 1992, 149, 590–598. [Google Scholar] [CrossRef]
- Mendes-Da-Cruz, D.A.; Belorio, E.P.; Cotta-De-Almeida, V. Thymus-Brain Connections in T-Cell Acute Lymphoblastic Leukemia. Neuroimmunomodulation 2024, 31, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Förster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Borroni, E.M.; Mantovani, A.; Locati, M.; Bonecchi, R. Chemokine receptors intracellular trafficking. Pharmacol. Ther. 2010, 127, 1–8. [Google Scholar] [CrossRef]
- Volpe, S.; Cameroni, E.; Moepps, B.; Thelen, S.; Apuzzo, T.; Thelen, M. CCR2 acts as scavenger for CCL2 during monocyte chemotaxis. PLoS ONE 2012, 7, e37208. [Google Scholar] [CrossRef]
- Bardi, G.; Lipp, M.; Baggiolini, M.; Loetscher, P. The T cell chemokine receptor CCR7 is internalized on stimulation with ELC, but not with SLC. Eur. J. Immunol. 2001, 31, 3291–3297. [Google Scholar] [CrossRef]
- Byers, M.A.; Calloway, P.A.; Shannon, L.; Cunningham, H.D.; Smith, S.; Li, F.; Fassold, B.C.; Vines, C.M. Arrestin 3 mediates endocytosis of CCR7 following ligation of CCL19 but not CCL21. J. Immunol. 2008, 181, 4723–4732. [Google Scholar] [CrossRef]
- Uetz-von Allmen, E.; Rippl, A.V.; Farhan, H.; Legler, D.F. A unique signal sequence of the chemokine receptor CCR7 promotes package into COPII vesicles for efficient receptor trafficking. J. Leukoc. Biol. 2018, 104, 375–389. [Google Scholar] [CrossRef]
- Correale, J.; Villa, A. Cellular elements of the blood-brain barrier. Neurochem. Res. 2009, 34, 2067–2077. [Google Scholar] [CrossRef]
- Shannon, L.A.; Calloway, P.A.; Welch, T.P.; Vines, C.M. CCR7/CCL21 migration on fibronectin is mediated by phospholipase Cgamma1 and ERK1/2 in primary T lymphocytes. J. Biol. Chem. 2010, 285, 38781–38787. [Google Scholar] [CrossRef]
- Bill, C.A.; Vines, C.M. Phospholipase C. Adv. Exp. Med. Biol. 2020, 1131, 215–242. [Google Scholar]
- Caers, J.; Peymen, K.; Suetens, N.; Temmerman, L.; Janssen, T.; Schoofs, L.; Beets, I. Characterization of G protein-coupled receptors by a fluorescence-based calcium mobilization assay. J. Vis. Exp. 2014, 28, e51516. [Google Scholar]
- Sestak, J.O.; Sullivan, B.P.; Thati, S.; Northrup, L.; Hartwell, B.; Antunez, L.; Forrest, M.L.; Vines, C.M.; Siahaan, T.J.; Berkland, C. Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis. Mol. Ther. Methods Clin. Dev. 2014, 1, 14008. [Google Scholar] [CrossRef] [PubMed]
- Badawi, A.H.; Kiptoo, P.; Wang, W.-T.; Choi, I.-Y.; Lee, P.; Vines, C.M.; Siahaan, T.J. Suppression of EAE and prevention of blood-brain barrier breakdown after vaccination with novel bifunctional peptide inhibitor. Neuropharmacology 2012, 62, 1874–1881. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19. [Google Scholar] [CrossRef]
- Kühn, R.; Schwenk, F.; Aguet, M.; Rajewsky, K. Inducible gene targeting in mice. Science 1995, 269, 1427–1429. [Google Scholar] [CrossRef]
- Flores-Montoya, M.G.; Bill, C.A.; Vines, C.M.; Sobin, C. Early chronic low-level lead exposure reduced C-C chemokine receptor 7 in hippocampal microglia. Toxicol. Lett. 2019, 314, 106–116. [Google Scholar] [CrossRef]
- Winter, S.S.; Sweatman, J.J.; Lawrence, M.B.; Rhoades, T.H.; Hart, A.L.; Larson, R.S. Enhanced T-lineage acute lymphoblastic leukaemia cell survival on bone marrow stroma requires involvement of LFA-1 and ICAM-1. Br. J. Haematol. 2001, 115, 862–871. [Google Scholar] [CrossRef]
- Puech, C.; Hodin, S.; Forest, V.; He, Z.; Mismetti, P.; Delavenne, X.; Perek, N. Assessment of HBEC-5i endothelial cell line cultivated in astrocyte conditioned medium as a human blood-brain barrier model for ABC drug transport studies. Int. J. Pharm. 2018, 551, 281–289. [Google Scholar] [CrossRef]
- Bolden, C.T.; Skibber, M.A.; Olson, S.D.; Rojas, M.Z.; Milewicz, S.; Gill, B.S.; Cox, C.S. Validation and characterization of a novel blood-brain barrier platform for investigating traumatic brain injury. Sci. Rep. 2023, 13, 16150. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardona, C.I.; Rodriguez, A.; Torres, V.C.; Sanchez, A.; Torres, A.; Vazquez, A.E.; Wagler, A.E.; Brissette, M.A.; Bill, C.A.; Vines, C.M. C-C Chemokine Receptor 7 Promotes T-Cell Acute Lymphoblastic Leukemia Invasion of the Central Nervous System via β2-Integrins. Int. J. Mol. Sci. 2024, 25, 9649. https://doi.org/10.3390/ijms25179649
Cardona CI, Rodriguez A, Torres VC, Sanchez A, Torres A, Vazquez AE, Wagler AE, Brissette MA, Bill CA, Vines CM. C-C Chemokine Receptor 7 Promotes T-Cell Acute Lymphoblastic Leukemia Invasion of the Central Nervous System via β2-Integrins. International Journal of Molecular Sciences. 2024; 25(17):9649. https://doi.org/10.3390/ijms25179649
Chicago/Turabian StyleCardona, Cesar I., Alondra Rodriguez, Vivian C. Torres, Anahi Sanchez, Angel Torres, Aaron E. Vazquez, Amy E. Wagler, Michael A. Brissette, Colin A. Bill, and Charlotte M. Vines. 2024. "C-C Chemokine Receptor 7 Promotes T-Cell Acute Lymphoblastic Leukemia Invasion of the Central Nervous System via β2-Integrins" International Journal of Molecular Sciences 25, no. 17: 9649. https://doi.org/10.3390/ijms25179649