

Computational Screening of T-Muurolol for an Alternative Antibacterial Solution against Staphylococcus aureus Infections: An In Silico Approach for Phytochemical-Based Drug Discovery

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Results
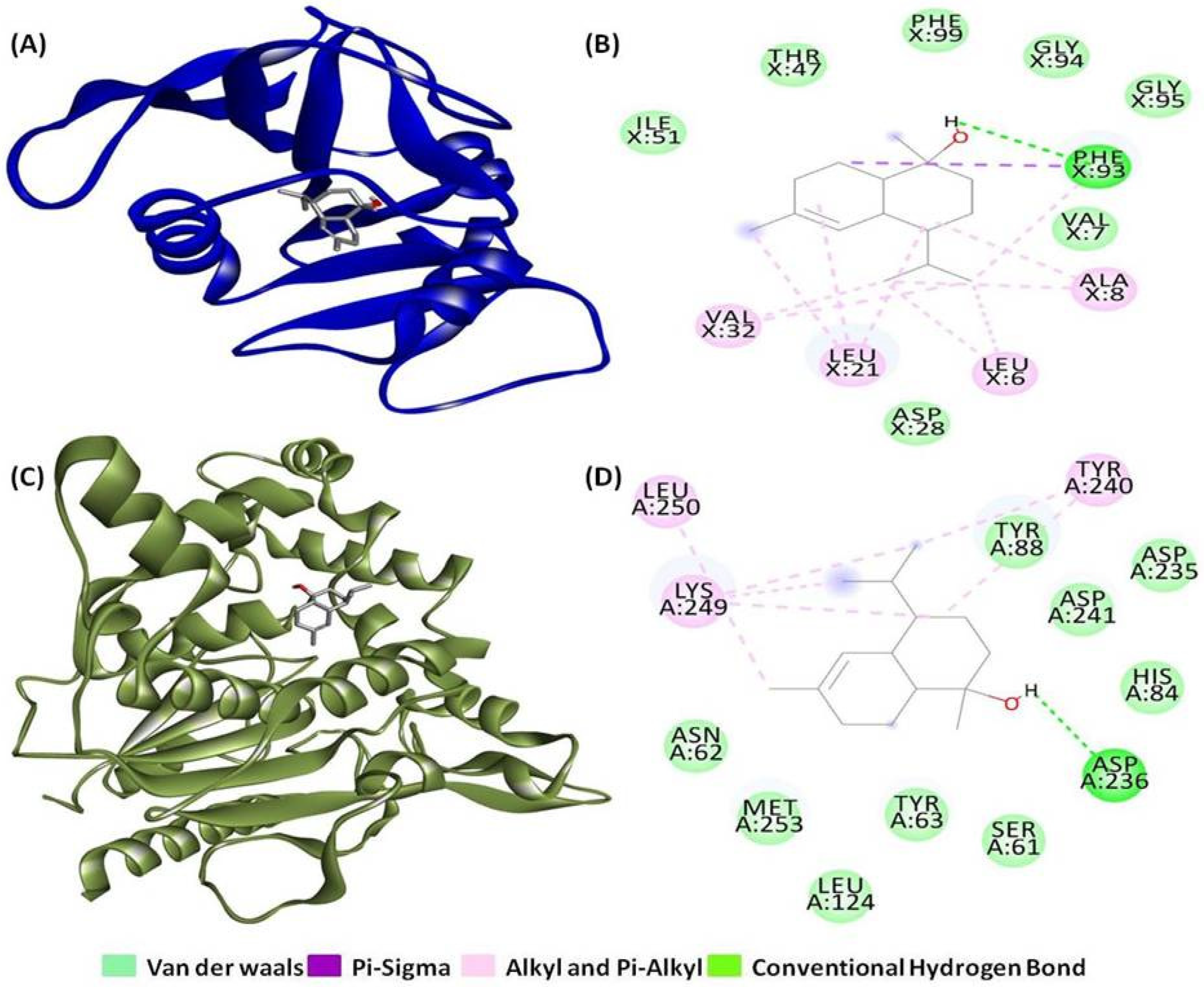
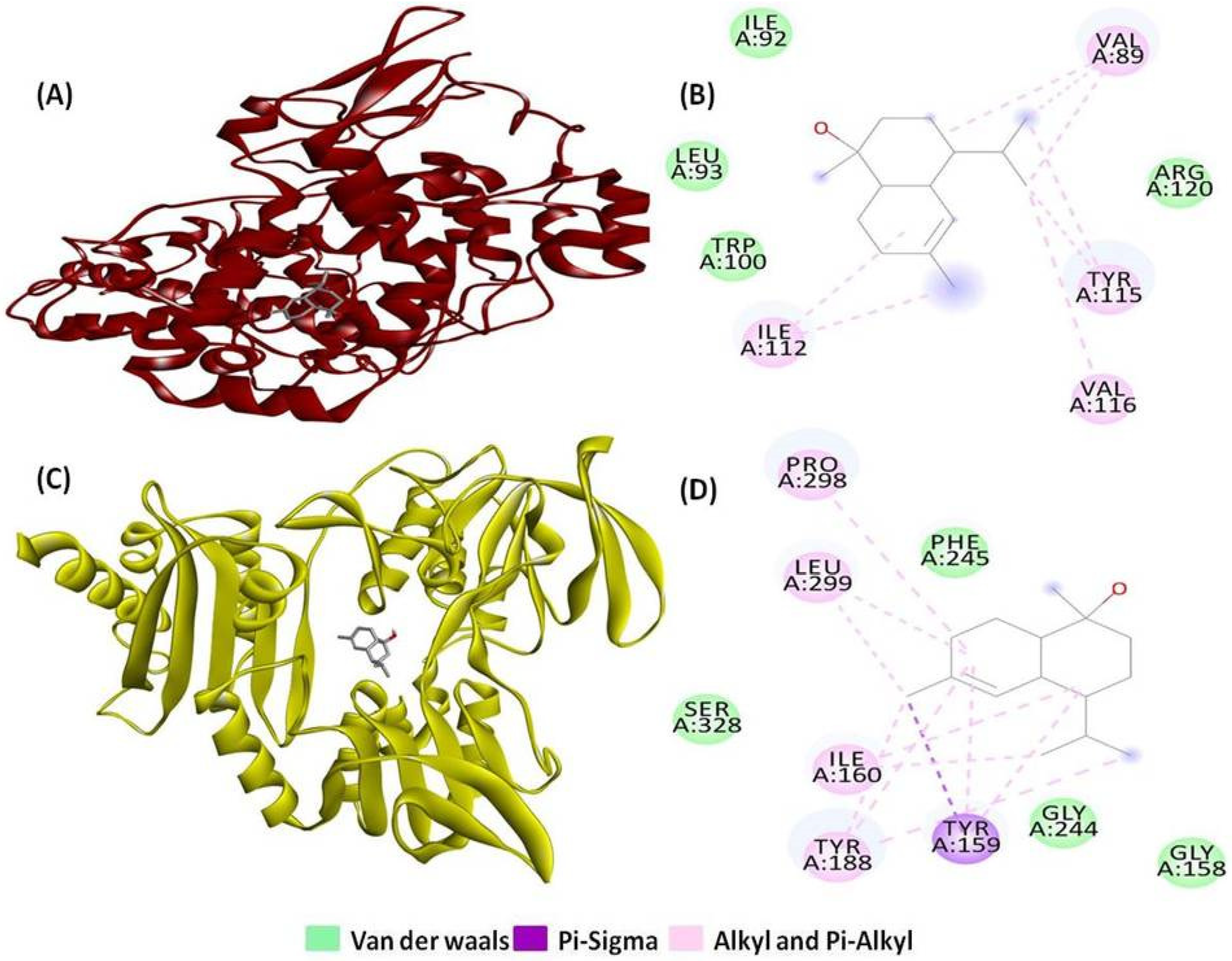
2.1. Molecular Docking and Interaction Analysis
2.2. ADMET Pharmacokinetic Analysis
2.2.1. Absorption
2.2.2. Distribution
2.2.3. Metabolism
2.2.4. Excretion
2.2.5. Toxicity
2.3. Drug-Likeness and Bio-Activity Analysis
2.4. MD Simulation Analysis
2.4.1. Monitoring Conformational Stability of the Backbone
2.4.2. Residual Flexibility Analysis
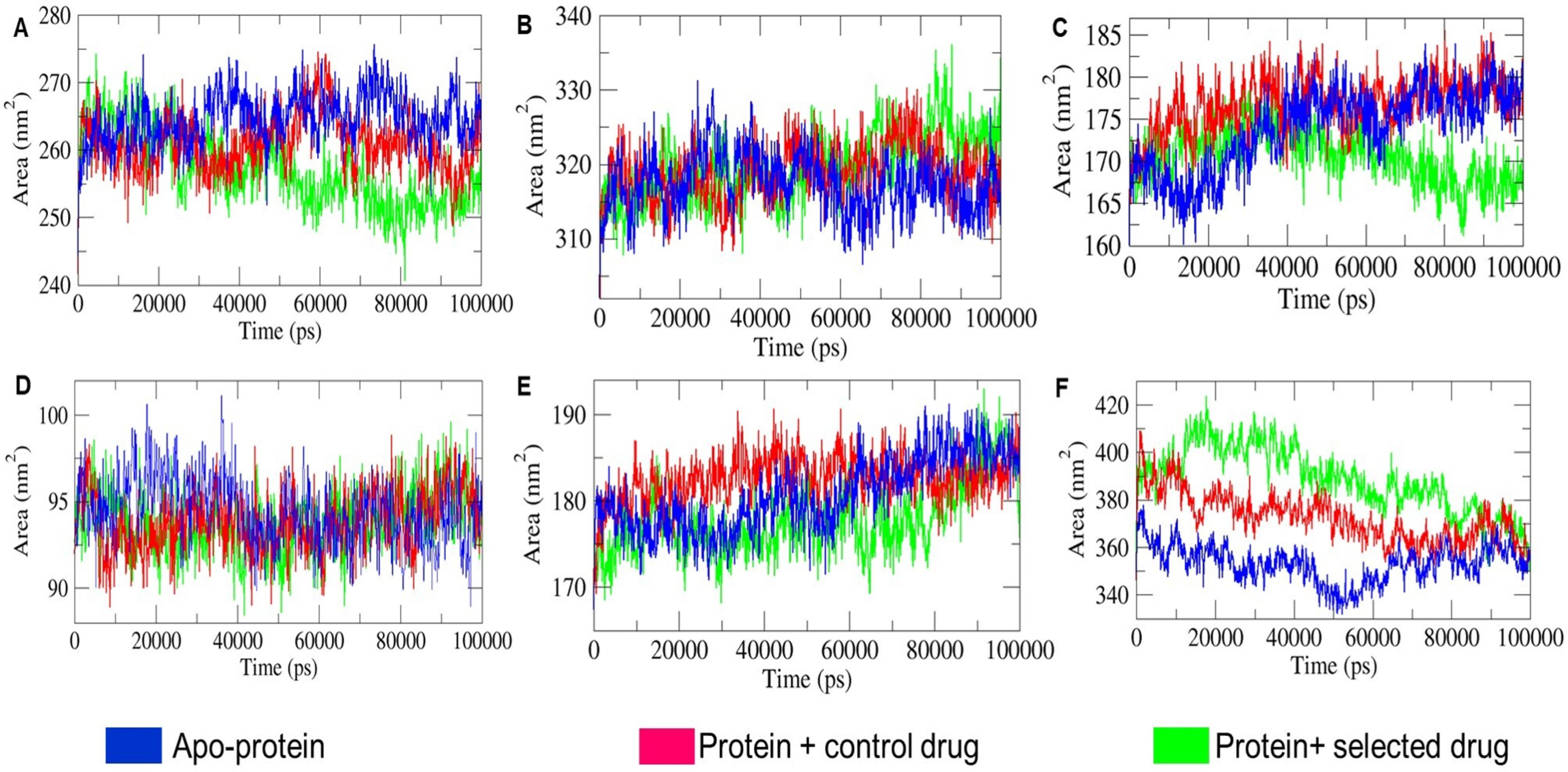
2.4.3. Structural Compactness Analysis
2.4.4. SASA Landscape Analysis
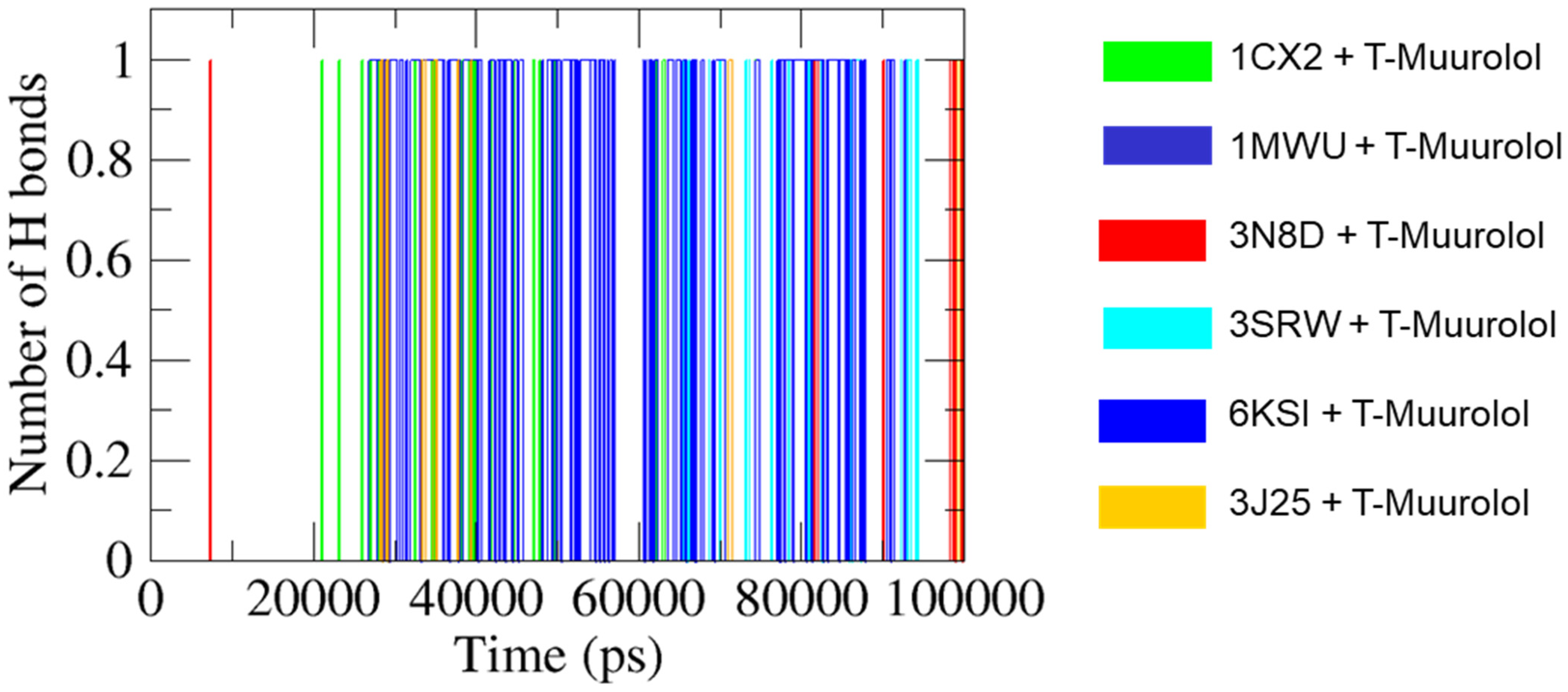
2.4.5. H Bonds Formation Analysis
2.5. Evaluation of MM-PBSA Binding Free Energy Decomposition
2.6. Quantum Chemical Computational Analysis
2.6.1. Frontier Molecular Orbital (FMO) Analysis
2.6.2. Global Chemical Reactivity Parameters
3. Discussion
4. Materials and Methods
4.1. Target Protein and Ligand Preparation
4.2. Molecular Docking and Binding Analysis
4.3. Pharmacokinetics Prediction and Bio-Activity Analysis
4.4. Molecular Dynamics and Simulation
4.5. Binding Free Energy Analysis
4.6. Computational Chemical Structure Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pollitt, E.J.G.; Szkuta, P.T.; Burns, N.; Foster, S.J. Staphylococcus aureus Infection Dynamics. PLoS Pathog. 2018, 14, e1007112. [Google Scholar] [CrossRef]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Liu, G.Y. Molecular Pathogenesis of Staphylococcus aureus Infection. Pediatr. Res. 2009, 65, 71–77. [Google Scholar] [CrossRef]
- Gatadi, S.; Madhavi, Y.V.; Chopra, S.; Nanduri, S. Promising Antibacterial Agents against Multidrug Resistant Staphylococcus aureus. Bioorg. Chem. 2019, 92, 103252. [Google Scholar] [CrossRef]
- Drew, R.H. Emerging Options for Treatment of Invasive, Multidrug-Resistant Staphylococcus aureus Infections. Pharmacotherapy 2007, 27, 227–249. [Google Scholar] [CrossRef]
- Shoaib, M.; Aqib, A.I.; Muzammil, I.; Majeed, N.; Bhutta, Z.A.; Kulyar, M.F.-A.; Fatima, M.; Zaheer, C.-N.F.; Muneer, A.; Murtaza, M.; et al. MRSA Compendium of Epidemiology, Transmission, Pathophysiology, Treatment, and Prevention within One Health Framework. Front. Microbiol. 2023, 13, 1067284. [Google Scholar] [CrossRef]
- Cosio, T.; Coniglione, F.; Flaminio, V.; Gaziano, R.; Coletta, D.; Petruccelli, R.; Dika, E.; Bianchi, L.; Campione, E. Pyodermitis during nivolumab treatment for non-small cell lung cancer: A case report and review of the literature. Int. J. Mol. Sci. 2023, 24, 4580. [Google Scholar] [CrossRef]
- Borges, A.; Saavedra, M.J.; Simoes, M. Insights on Antimicrobial Resistance, Biofilms and the Use of Phytochemicals as New Antimicrobial Agents. Curr. Med. Chem. 2015, 22, 2590–2614. [Google Scholar] [CrossRef]
- Dhama, K.; Tiwari, R.; Chakraborty, S.; Saminathan, M.; Kumar, A.; Karthik, K.; Wani, M.Y.; Amarpal, A.; Singh, S.; Rahal, A. Evidence Based Antibacterial Potentials of Medicinal Plants and Herbs Countering Bacterial Pathogens Especially in the Era of Emerging Drug Resistance: An Integrated Update. Int. J. Pharmacol. 2014, 10, 1–43. [Google Scholar] [CrossRef]
- Solanki, R.; Jodha, B.; Prabina, K.E.; Aggarwal, N.; Patel, S. Recent Advances in Phytochemical Based Nano-Drug Delivery Systems to Combat Breast Cancer: A Review. J. Drug Deliv. Sci. Technol. 2022, 77, 103832. [Google Scholar] [CrossRef]
- Egbuna, C.; Kumar, S.; Ifemeje, J.C.; Ezzat, S.M.; Kaliyaperumal, S. Phytochemicals as Lead Compounds for New Drug Discovery; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 978-0-12-817891-1. [Google Scholar]
- Suganya, T.; Packiavathy, I.A.S.V.; Aseervatham, G.S.B.; Carmona, A.; Rashmi, V.; Mariappan, S.; Devi, N.R.; Ananth, D.A. Tackling Multiple-Drug-Resistant Bacteria with Conventional and Complex Phytochemicals. Front. Cell. Infect. Microbiol. 2022, 12, 883839. [Google Scholar] [CrossRef] [PubMed]
- Elzaawely, A.A.; Xuan, T.D.; Koyama, H.; Tawata, S. Antioxidant Activity and Contents of Essential Oil and Phenolic Compounds in Flowers and Seeds of Alpinia zerumbet (Pers.) B.L. Burtt. & R.M. Sm. Food Chem. 2007, 104, 1648–1653. [Google Scholar] [CrossRef]
- Cheng, S.-S.; Wu, C.-L.; Chang, H.-T.; Kao, Y.-T.; Chang, S.-T. Antitermitic and Antifungal Activities of Essential Oil of Calocedrus formosana Leaf and Its Composition. J. Chem. Ecol. 2004, 30, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Sahingil, D. GC/MS-Olfactometric Characterization of the Volatile Compounds, Determination Antimicrobial and Antioxidant Activity of Essential Oil from Flowers of Calendula (Calendula officinalis L.). J. Essent. Oil Bear. Plants 2019, 22, 1571–1580. [Google Scholar] [CrossRef]
- Chandra, A.; Chaudhary, M.; Qamar, I.; Singh, N.; Nain, V. In Silico Identification and Validation of Natural Antiviral Compounds as Potential Inhibitors of SARS-CoV-2 Methyltransferase. J. Biomol. Struct. Dyn. 2022, 40, 6534–6544. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Nandi, R.; Vishwakarma, P.; Prakash, A.; Kumar, D. Discovering Potential RNA Dependent RNA Polymerase Inhibitors as Prospective Drugs against COVID-19: An In Silico Approach. Front. Pharmacol. 2021, 12, 634047. [Google Scholar] [CrossRef]
- Bhowmik, D.; Nandi, R.; Jagadeesan, R.; Kumar, N.; Prakash, A.; Kumar, D. Identification of Potential Inhibitors against SARS-CoV-2 by Targeting Proteins Responsible for Envelope Formation and Virion Assembly Using Docking Based Virtual Screening, and Pharmacokinetics Approaches. Infect. Genet. Evol. 2020, 84, 104451. [Google Scholar] [CrossRef]
- Sinha, M.; Jagadeesan, R.; Kumar, N.; Saha, S.; Kothandan, G.; Kumar, D. In-Silico Studies on Myo Inositol-1-Phosphate Synthase of Leishmania donovani in Search of Anti-Leishmaniasis. J. Biomol. Struct. Dyn. 2022, 40, 3371–3384. [Google Scholar] [CrossRef]
- Abdelrheem, D.A.; Rahman, A.A.; Elsayed, K.N.M.; Abd El-Mageed, H.R.; Mohamed, H.S.; Ahmed, S.A. Isolation, Characterization, In Vitro Anticancer Activity, Dft Calculations, Molecular Docking, Bioactivity Score, Drug-Likeness and Admet Studies of Eight Phytoconstituents from Brown Alga Sargassum platycarpum. J. Biomol. Struct. Dyn. 2021, 1225, 129245. [Google Scholar] [CrossRef]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Chaudhary, N.; Aparoy, P. Deciphering the Mechanism behind the Varied Binding Activities of COXIBs through Molecular Dynamic Simulations, MM-PBSA Binding Energy Calculations and per-Residue Energy Decomposition Studies. J. Biomol. Struct. Dyn. 2017, 35, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Kesavan, A.K.; Juneja, V.; Thakur, S. Molecular Modeling, Simulation and Docking of Rv1250 Protein from Mycobacterium Tuberculosis. Front. Bioinform. 2023, 3, 1125479. [Google Scholar] [CrossRef]
- Arooj, M.; Shehadi, I.; Nassab, C.N.; Mohamed, A.A. Physicochemical Stability Study of Protein–Benzoic Acid Complexes Using Molecular Dynamics Simulations. Amino Acids 2020, 52, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated Interaction Energy (SIE) for Scoring Protein−Ligand Binding Affinities. 1. Exploring the Parameter Space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Kumar, A.; Debnath, M. Molecular Docking and Simulation Studies to Give Insight of Surfactin Amyloid Interaction for Destabilizing Alzheimer’s Aβ42 Protofibrils. Med. Chem. Res. 2016, 25, 1616–1622. [Google Scholar] [CrossRef]
- Koopmans, T. Über Die Zuordnung von Wellenfunktionen Und Eigenwerten Zu Den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Assaeed, A.; Elshamy, A.; El Gendy, A.E.-N.; Dar, B.; Al-Rowaily, S.; Abd-ElGawad, A. Sesquiterpenes-Rich Essential Oil from Above Ground Parts of Pulicaria somalensis Exhibited Antioxidant Activity and Allelopathic Effect on Weeds. Agronomy 2020, 10, 399. [Google Scholar] [CrossRef]
- Górski, K.M.; Kowalczyk, T.; Picot, L.; Rijo, P.; Ghorbanpour, M.; Sitarek, P. The Precious Potential of the Sacred Tree Chamaecyparis obtusa (Siebold & Zucc.) Endl. as a Source of Secondary Metabolites with Broad Biological Applications. Int. J. Mol. Sci. 2024, 25, 2723. [Google Scholar] [CrossRef]
- Radulovic, N.S.; Blagojevic, P.D.; Stojanovic-Radic, Z.Z.; Stojanovic, N.M. Antimicrobial plant metabolites: Structural diversity and mechanism of action. Curr. Med. Chem. 2013, 20, 932–952. [Google Scholar]
- Hu, C.; Xiong, N.; Zhang, Y.; Rayner, S.; Chen, S. Functional Characterization of Lipase in the Pathogenesis of Staphylococcus aureus. Biochem. Biophys. Res. Commun. 2012, 419, 617–620. [Google Scholar] [CrossRef]
- Jianu, C.; Stoin, D.; Cocan, I.; David, I.; Pop, G.; Lukinich-Gruia, A.T.; Mioc, M.; Mioc, A.; Șoica, C.; Muntean, D.; et al. In silico and in vitro evaluation of the antimicrobial and antioxidant potential of Mentha × smithiana R. Graham essential oil from Western Romania. Foods 2021, 10, 815. [Google Scholar] [CrossRef] [PubMed]
- Dönhöfer, A.; Franckenberg, S.; Wickles, S.; Berninghausen, O.; Beckmann, R.; Wilson, D.N. Structural Basis for TetM-Mediated Tetracycline Resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 16900–16905. [Google Scholar] [CrossRef]
- Cipriano, A.; Viviano, M.; Feoli, A.; Milite, C.; Sarno, G.; Castellano, S.; Sbardella, G. NADPH Oxidases: From Molecular Mechanisms to Current Inhibitors. J. Med. Chem. 2023, 66, 11632–11655. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Chemical Hardness and Density Functional Theory. J. Chem. Sci. 2005, 117, 369–377. [Google Scholar] [CrossRef]
- Klingmüller, U.; Schilling, M.; Depner, S.; D’Alessandro, L. Biological Foundations of Signal Transduction, Systems Biology and Aberrations in Disease. In Computational Systems Biology: From Molecular Mechanisms to Disease, 2nd ed.; Academic Press: Amsterdam, The Netherlands, 2014; pp. 45–64. ISBN 978-0-12-405926-9. [Google Scholar]
- Haroon, M.; Akhtar, T.; Shaikh, Q.; Mehmood, H.; Khalid, M.; Asghar, M.A.; Alshehri, S.M.; Ojha, S.C. Facile Synthesis and DFT Analysis of Novel Thiazole-Based Hydrazones: An Experimental and Theoretical Perspective. ACS Omega 2023, 8, 27488–27499. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Hsu, K.P.; Wang, E.I.C.; Ho, C.L. Composition, in vitro cytotoxic, and antimicrobial activities of the flower essential oil of Diospyros discolor from Taiwan. Nat. Prod. Commun. 2015, 10, 1934578X1501000744. [Google Scholar] [CrossRef]
- Zouaghi, N.; Bensiradj, N.H.; Cavaleiro, C.; Nadjemi, B.; Trari, M. Phytochemical study and antibacterial effects of Fraxinus angustifolia Vahl (Algeria): Experimental and computational investigations. Waste Biomass Valor. 2021, 12, 3605–3616. [Google Scholar] [CrossRef]
- Khoury, M.; El Beyrouthy, M.; Ouaini, N.; Eparvier, V.; Stien, D. Hirtellina lobelii DC. essential oil, its constituents, its combination with antimicrobial drugs and its mode of action. Fitoterapia 2019, 133, 130–136. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Dutta, A.; Khanra, P.K.; Gupta, N.; Dutta, R.; Tzvetkov, N.T.; Milella, L.; Ponticelli, M. In silico exploration of 4 (α-l-rhamnosyloxy)-benzyl isothiocyanate: A promising phytochemical-based drug discovery approach for combating multi-drug resistant Staphylococcus aureus. Comput. Biol. Med. 2024, 179, 108907. [Google Scholar] [CrossRef]
- Gupta, N.; Bhattacharya, S.; Urbanová, K.; Dutta, A.; Hazra, A.K.; Fernández-Cusimamani, E.; Leuner, O. Systematic Analysis of Antimicrobial Activity, Phytochemistry, and in Silico Molecular Interaction of Selected Essential Oils and Their Formulations from Different Indian Spices against Foodborne Bacteria. Heliyon 2023, 9, e22480. [Google Scholar] [CrossRef]
- Kandasamy, T.; Sen, P.; Ghosh, S.S. Multi-Targeted Drug Repurposing Approach for Breast Cancer via Integrated Functional Network Analysis. Mol. Inform. 2022, 41, 2100300. [Google Scholar] [CrossRef] [PubMed]
- Gaussian 09 Citation. Available online: https://gaussian.com/g09citation/ (accessed on 28 May 2024).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contents | PDB Entry | Ligands | |||
|---|---|---|---|---|---|
| T-Muurolol | Ascorbic Acid | Ibuprofen | 2-Oxazolidinone | ||
| Anti-inflammatory Proteins | 4YYZ | −5.7 | - | −6.2 | - |
| 1N8Q | −6.5 | - | −7.7 | - | |
| 2PE4 | −5.8 | - | −6.2 | - | |
| 3V92 | −6.7 | - | −6.3 | - | |
| 4CX7 | −5.7 | - | −5.7 | - | |
| 1CX2 | −6.3 | - | −6.4 | - | |
| Antioxidant Proteins | 1HCK | −6.2 | −4.8 | - | - |
| 2CDU | −6.4 | −5.4 | - | - | |
| 2F8A | −4.9 | −4.4 | - | - | |
| 3HFF | −4.3 | −4.1 | - | - | |
| SA Pathogenesis Proteins | 2O8L | −6.2 | - | - | −3.4 |
| 4AE5 | −5.7 | - | −3 | ||
| 6KSI | −7.1 | - | - | −3.9 | |
| Universal Bacterial Proteins for their Survival | 1JZQ | −7.2 | - | - | −3.6 |
| 1KZN | −6.1 | - | - | −3.7 | |
| 2VEG | −5.3 | - | - | −3.9 | |
| 2ZDQ | −5.8 | - | - | −3.9 | |
| 3RAE | −6 | - | - | −4 | |
| 3SRW | −7.5 | - | - | −3.7 | |
| 3UDI | −5.8 | - | - | −3.5 | |
| 3TTZ | −5.9 | - | - | −3.3 | |
| MRSA | 1GHI | −6 | - | - | −3.3 |
| 1MWU | −6.1 | - | - | −3.6 | |
| VRSA | 3N8D | −5.9 | - | - | −3.9 |
| TetRSA | 2FJ1 | −5.7 | - | - | −3.5 |
| 3J25 | −6.6 | - | - | −3.3 | |
| 7Y58 | −5.8 | - | - | −3.7 | |
| PDB Entry | No. of H-Bonds | H-Bonds and Interacting Residues | No. of Other Interactions | Other Interactions and Numbers | Other Interaction and Interacting Residues |
|---|---|---|---|---|---|
| 1CX2 | 0 | 8 | Alkyl and Pi-alkyl (8) | Ile112 (2), Tyr115 (2), Val89 (3), Val116 (1) | |
| 1MWU | 1 | Glu294 (1) | 6 | Alkyl and Pi-alkyl (5), Pi-sigma (1) | Ala276 (1), Lys289 (3), Tyr272 (2) |
| 2CDU | 0 | 11 | Alkyl and Pi-alkyl (10), Pi-sigma (1) | Ile160 (2), Leu299 (2), Pro298 (1), Tyr159 (3), Tyr188 (3) | |
| 3J25 | 0 | 10 | Alkyl and Pi-alkyl (10) | Ala554 (2), Cys603 (2), Ile544 (2), Leu593 (4) | |
| 3N8D | 0 | 2 | Alkyl (2) | Val19 (2) | |
| 3SRW | 1 | Phe93 (1) | 11 | Alkyl and Pi-alkyl (10), Pi-sigma (1) | Ala8 (2), Leu6 (2), Leu21 (3), Phe93 (2), Val32 (2) |
| 6KSI | 1 | Asp236 (1) | 6 | Alkyl and Pi-alkyl (6) | Leu250 (1), Lys249 (3), Tyr240 (2) |
| Property | Model Name | Compounds | ||
|---|---|---|---|---|
| T-Muurolol | Ibuprofen | 2-Oxazolidinone | ||
| Absorption | Water solubility (log mol/L) | −4.073 | −3.696 | 0.243 |
| Caco2 permeability (log Papp in 10−6 cm/s) | 1.479 | 1.729 | 1.582 | |
| GI absorption (%) | 94.296 | 94.064 | 98.901 | |
| P-glycoprotein substrate | No | No | No | |
| P-glycoprotein I inhibitor | No | No | No | |
| P-glycoprotein II inhibitor | No | No | No | |
| Distribution | VDss (human) (log L/kg) | 0.42 | −0.803 | −0.092 |
| Fraction unbound (human) | 0.28 | 0.239 | 0.813 | |
| Metabolism | CYP2D6 substrate | No | No | No |
| CYP3A4 substrate | No | No | No | |
| CYP1A2 inhibitor | No | No | No | |
| CYP2C19 inhibitor | No | No | No | |
| CYP2C9 inhibitor | No | No | No | |
| CYP2D6 inhibitor | No | No | No | |
| CYP3A4 inhibitor | No | No | No | |
| Excretion | Total clearance (log mL/min/kg) | 1.085 | 0.263 | 0.496 |
| Renal OCT2 substrate | No | No | No | |
| Toxicity | AMES toxicity | No | No | Yes |
| Max. tolerated dose (human) (log mg/kg/day) | 0.343 | 1.015 | 1.249 | |
| hERG I inhibitor | No | No | No | |
| hERG II inhibitor | No | No | No | |
| Oral Rat Acute Toxicity (LD50) (mol/kg) | 1.918 | 2.303 | 2.084 | |
| Hepatotoxicity | No | Yes | No | |
| Skin Sensitisation | Yes | Yes | No | |
| Minnow toxicity (log mM) | 0.743 | 0.619 | 2.872 | |
| Compounds | Lipinski’s Rule of Five | Molecular Weight (g/mol) | H-Bond Acceptors | H-Bond Donors | LogP | TPSA (Å2) | Bioavailability Score |
|---|---|---|---|---|---|---|---|
| T-muurolol | Yes; zero violation | 222.37 | 1 | 1 | 3.42 | 20.23 | 0.55 |
| Ibuprofen | Yes; zero violation | 206.28 | 2 | 1 | 3.01 | 37.3 | 0.85 |
| 2-Oxazolidinone | Yes; zero violation | 87.08 | 2 | 1 | −0.04 | 38.33 | 0.55 |
| Compounds | * GPCRs | ICM | KI | NRL | PI | EI |
|---|---|---|---|---|---|---|
| T-muurolol | −0.09 | 0.05 | −0.87 | 0.39 | −0.63 | 0.4 |
| Ibuprofen | −0.17 | −0.01 | −0.72 | 0.05 | −0.21 | 0.12 |
| 2-Oxazolidinone | −3.47 | −3.26 | −3.67 | −3.78 | −3.23 | −3.4 |
| Macromolecules | Van der Waals Energy (KJ/mol) | Electrostatic Energy (KJ/mol) | Polar Solvation Energy (KJ/mol) | SASA Energy (KJ/mol) | Binding Free Energy (KJ/mol) |
|---|---|---|---|---|---|
| 1CX2 | −53.91 | −2.68 | 23.93 | −6.82 | −39.48 |
| 1MWU | −0.05 | 0.46 | −55.05 | −0.93 | −55.58 |
| 3N8D | −12.92 | 0.58 | −6.58 | −2.46 | −21.39 |
| 3SRW | −29.99 | −1.68 | 24.85 | −4.21 | −11.03 |
| 6KSI | −71.82 | −4.36 | 36.27 | −10.72 | −50.64 |
| 3J25 | −5.164 | −0.14 | 2.14 | −1.84 | −5.01 |
| List of Parameters | Values Calculated at the DFT Level |
|---|---|
| Dipole moment (in debye or D) | 1.67 |
| EHOMO (in eV) | −6.37 |
| ELUMO (in eV) | 0.52 |
| electronegativity index (χ, in eV) | 2.92 |
| chemical potential index (μ, in eV) | −2.92 |
| global hardness index (η, in eV) | 3.45 |
| softness index (σ, in eV−1) | 0.29 |
| global electrophilicity index (ω, in eV) | 1.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, S.; Khanra, P.K.; Dutta, A.; Gupta, N.; Aliakbar Tehrani, Z.; Severová, L.; Šrédl, K.; Dvořák, M.; Fernández-Cusimamani, E. Computational Screening of T-Muurolol for an Alternative Antibacterial Solution against Staphylococcus aureus Infections: An In Silico Approach for Phytochemical-Based Drug Discovery. Int. J. Mol. Sci. 2024, 25, 9650. https://doi.org/10.3390/ijms25179650
Bhattacharya S, Khanra PK, Dutta A, Gupta N, Aliakbar Tehrani Z, Severová L, Šrédl K, Dvořák M, Fernández-Cusimamani E. Computational Screening of T-Muurolol for an Alternative Antibacterial Solution against Staphylococcus aureus Infections: An In Silico Approach for Phytochemical-Based Drug Discovery. International Journal of Molecular Sciences. 2024; 25(17):9650. https://doi.org/10.3390/ijms25179650
Chicago/Turabian StyleBhattacharya, Soham, Pijush Kanti Khanra, Adrish Dutta, Neha Gupta, Zahra Aliakbar Tehrani, Lucie Severová, Karel Šrédl, Marek Dvořák, and Eloy Fernández-Cusimamani. 2024. "Computational Screening of T-Muurolol for an Alternative Antibacterial Solution against Staphylococcus aureus Infections: An In Silico Approach for Phytochemical-Based Drug Discovery" International Journal of Molecular Sciences 25, no. 17: 9650. https://doi.org/10.3390/ijms25179650
APA StyleBhattacharya, S., Khanra, P. K., Dutta, A., Gupta, N., Aliakbar Tehrani, Z., Severová, L., Šrédl, K., Dvořák, M., & Fernández-Cusimamani, E. (2024). Computational Screening of T-Muurolol for an Alternative Antibacterial Solution against Staphylococcus aureus Infections: An In Silico Approach for Phytochemical-Based Drug Discovery. International Journal of Molecular Sciences, 25(17), 9650. https://doi.org/10.3390/ijms25179650