
Predictive Biomarkers and Resistance Mechanisms of Checkpoint Inhibitors in Malignant Solid Tumors
 , ,
, ,  and
and
Abstract
1. Introduction
2. Immune Checkpoint Inhibition in Cancer—Mechanisms of Action and Resistance Mechanisms
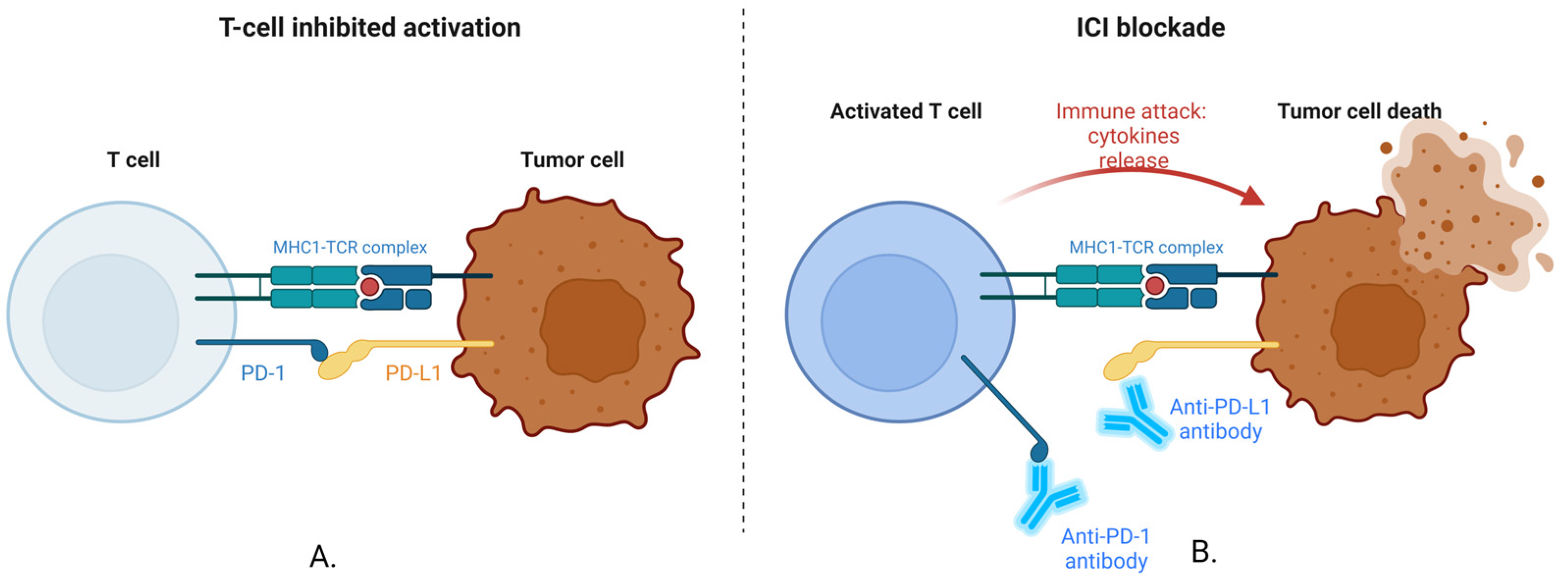
2.1. Blocking PD-1/PD-L1 Interaction
2.2. Blocking CTLA-4 Interaction
2.3. Enhanced T-Cell Activation and Proliferation
2.4. Induction of Immunological Memory
2.5. Reactivation of Exhausted T Cells
2.6. Overcoming Tumor-Induced Immunosuppression
2.7. Synergy with Other Immune Cells
- Inhibitory immune checkpoints beyond PD-1/PD-L1 and CTLA-4: Other immune checkpoint molecules that can similarly suppress T-cell function, such as TIM-3, LAG-3, or VISTA, may also be expressed by tumors [66,67]. If these other pathways are activated, it might not be enough to use ICIs that only target CTLA-4 or PD-1/PD-L1 [66,67].
- After a first response to the therapy, acquired (secondary) resistance arises, which ultimately results in treatment failure [70]. It can arise from a variety of mechanisms, including signaling pathway mutations, decreased neoantigen expression, upregulation of alternative immune checkpoints, elevated PD-L1 expression, T-cell exhaustion, metabolic changes in the tumor microenvironment, or immunosuppressive cell recruitment [71].
- Mutations in signaling pathways: Signaling pathway abnormalities in tumor cells can enable them to evade immune recognition [72,73,74]. A tumor’s capacity to react to interferon-gamma (IFN-γ), a cytokine essential for immune-mediated tumor elimination, can be hindered by mutations in the JAK1/2 gene, for instance [72,73,74].
3. Exploratory Biomarkers for Immune Checkpoint Inhibition
4. Approved Biomarkers for Immune Checkpoint Inhibition
4.1. MSI or dMMR
4.2. TMB
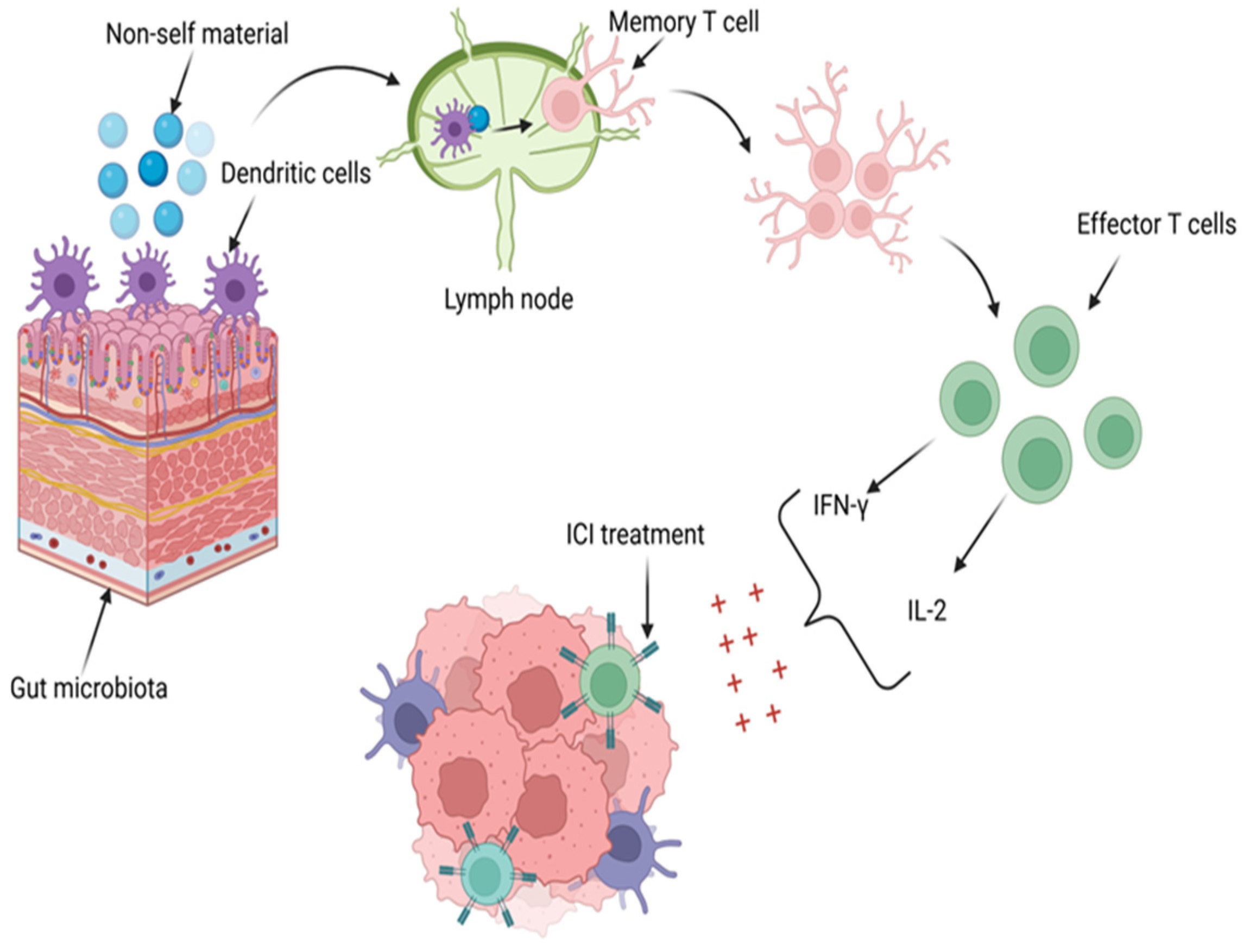
5. Microbiota as an Immune Adjuvant for Immune Checkpoint Blockade
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| APCs | antigen-presenting cells |
| B2M | beta-2-microglobulin |
| CRC | colorectal cancer |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| DDR | DNA damage response |
| DNA | deoxyribonucleic acid |
| dMMR | defective DNA mismatch repair |
| EMA | European Medicines Agency |
| ER | estrogen receptor |
| EU | European Union |
| EpCAM | epithelial cell adhesion molecule |
| FDA | Food and Drug Administration |
| GITR | glucocorticoid-induced TNFR-related protein |
| HCC | hepatocellular carcinoma |
| HER2 | human epidermal growth factor receptor 2 |
| HLA-DR | human leukocyte antigen-DR isotype |
| HNPCC | Hereditary Non-Polyposis Colorectal Cancer |
| ICI | immune checkpoint inhibitor |
| IFN-γ | interferon-gamma |
| Ig | immunoglobulin |
| IL-10 | interleukin 10 |
| irAEs | immune-related adverse events |
| IT | immunotherapy |
| ITIM | immunoreceptor tyrosine-based inhibitory motif |
| JAK1/2 | Janus kinases |
| KEAP 1 | Kelch-like ECH-associated protein 1 |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LAG-3 | lymphocyte activation gene-3 |
| LS | Lynch syndrome |
| MDSCs | myeloid-derived suppressor cells |
| MET | mesenchymal–epithelial transition |
| MHC | major histocompatibility complex |
| MSI | microsatellite instability |
| MSI-H | microsatellite instability-high |
| NK | natural killer |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| NSCLC | non-small cell lung cancer |
| NTRK | neurotrophic tyrosine receptor kinase |
| PD-1 | programmed cell death 1 |
| PD-L1 | programmed cell death ligand 1 |
| PDT | photodynamic therapy |
| PMBCL | primary mediastinal large B-cell lymphoma |
| POLE | polymerase epsilon |
| RCC | renal cell carcinoma |
| RET | rearranged during transfection |
| RT | radiotherapy |
| SCLC | small cell lung cancer |
| SHP-1, SHP-2 | protein tyrosine phosphatase 1 and 2 |
| STK11 | serine/threonine kinase 11 |
| STK11/LKB1 | STK11 gene codes for liver kinase B1 |
| TAMs | tumor-associated macrophages |
| TCMs | memory T cells |
| TCR | T-cell receptor |
| TGF-β | transforming growth factor beta |
| Th | helper T cell |
| TIGIT | T-cell immunoglobulin and ITIM domain |
| TILs | tumor-infiltrating T cells |
| TIM-3 | T-cell immunoglobulin and mucin domain-containing protein 3 |
| TMB | tumor mutational burden |
| TML | tumor mutational load |
| TME | tumor microenvironment |
| TNBC | triple-negative breast cancer |
| TNF-α | tumor necrosis factor-α |
| TPEX | exhausted T cells and their precursors |
| TRM | tissue-resident memory T cells |
| Tregs | regulatory T cells |
| UC | urothelial carcinoma |
| USA | United States of America |
| VEGF | vascular endothelial growth factor |
| VISTA | V-domain Ig suppressor of T-cell activation |
References
- Fujiwara, Y.; Mittra, A.; Naqash, A.R.; Takebe, N. A review of mechanisms of resistance to immune checkpoint inhibitors and potential strategies for therapy. Cancer Drug Resist. 2020, 3, 252–275. [Google Scholar] [CrossRef] [PubMed]
- Basudan, A.M. The Role of Immune Checkpoint Inhibitors in Cancer Therapy. Clin. Pract. 2022, 13, 22–40. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, P.; Cohn, M. A Theory of self-nonself discrimination. Science 1970, 169, 1042–1049. [Google Scholar] [CrossRef]
- Jenkins, M.K.; Schwartz, R.H. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 1987, 165, 302–319. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Linhares, A.; Leitner, J.; Grabmeier-Pfistershammer, K.; Steinberger, P. Not All Immune Checkpoints Are Created Equal. Front. Immunol. 2018, 9, 1909. [Google Scholar] [CrossRef]
- Sambi, M.; Bagheri, L.; Szewczuk, M.R. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. J. Oncol. 2019, 2019, 45087941. [Google Scholar] [CrossRef]
- Koustas, E.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M.V. The Resistance Mechanisms of Checkpoint Inhibitors in Solid Tumors. Biomolecules 2020, 10, 666. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Shum, B.; Larkin, J.; Turajlic, S. Predictive biomarkers for response to immune checkpoint inhibition. Semin. Cancer Biol. 2021, 79, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.V.; Whittington, R.; Malkowicz, S.B.; Schultz, D.; Blank, K.; Broderick, G.A.; Tomaszewski, J.E.; Renshaw, A.A.; Kaplan, I.; Beard, C.J.; et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 1998, 280, 969–974. [Google Scholar] [CrossRef]
- Routy, B.; le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Elkrief, A.; Derosa, L.; Zitvogel, L.; Kroemer, G.; Routy, B. The intimate relationship between gut microbiota and cancer immunotherapy. Gut Microbes 2019, 10, 424–428. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Alturki, N.A. Review of the Immune Checkpoint Inhibitors in the Context of Cancer Treatment. J. Clin. Med. 2023, 12, 4301. [Google Scholar] [CrossRef] [PubMed]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Burns, M.C.; O’donnell, A.; Puzanov, I. Pembrolizumab for the treatment of advanced melanoma. Expert Opin. Orphan Drugs 2016, 4, 867–873. [Google Scholar] [CrossRef]
- Raedler, L.A. Opdivo (Nivolumab): Second PD-1 Inhibitor Receives FDA Approval for Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 180–183. [Google Scholar] [PubMed]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.F.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Alkholifi, F.K.; Alsaffar, R.M. Dostarlimab an Inhibitor of PD-1/PD-L1: A New Paradigm for the Treatment of Cancer. Medicina 2022, 58, 1572. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, N.; Wang, X.; Ravindranathan, S.; Chin, D.J.; Tseng, S.-Y.; Klakamp, S.L.; Widmann, K.; Kapoor, V.N.; Vexler, V.; Keegan, P.; et al. Toripalimab, a therapeutic monoclonal anti-PD-1 antibody with high binding affinity to PD-1 and enhanced potency to activate human T cells. Cancer Immunol. Immunother. 2024, 73, 60. [Google Scholar] [CrossRef] [PubMed]
- Suzman, D.L.; Agrawal, S.; Ning, Y.-M.; Maher, V.E.; Fernandes, L.L.; Karuri, S.; Tang, S.; Sridhara, R.; Schroeder, J.; Goldberg, K.B.; et al. FDA Approval Summary: Atezolizumab or Pembrolizumab for the Treatment of Patients with Advanced Urothelial Carcinoma Ineligible for Cisplatin-Containing Chemotherapy. Oncologist 2018, 24, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Spira, A.; Raben, D.; Planchard, D.; Cho, B.; Özgüroğlu, M.; Daniel, D.; Villegas, A.; Vicente, D.; Hui, R.; et al. Outcomes with durvalumab by tumour PD-L1 expression in unresectable, stage III non-small-cell lung cancer in the PACIFIC trial. Ann. Oncol. 2020, 31, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Avelumab: A Review in Metastatic Merkel Cell Carcinoma. Target. Oncol. 2018, 13, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Mansh, M. Ipilimumab and cancer immunotherapy: A new hope for advanced stage melanoma. Yale J. Biol. Med. 2011, 84, 381–389. [Google Scholar]
- Keam, S.J. Tremelimumab: First Approval. Drugs 2023, 83, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Sheng, I.Y.; Ornstein, M.C. Ipilimumab and Nivolumab as First-Line Treatment of Patients with Renal Cell Carcinoma: The Evidence to Date. Cancer Manag. Res. 2020, 12, 4871–4881. [Google Scholar] [CrossRef] [PubMed]
- Mohsenzadegan, M.; Bavandpour, P.; Nowroozi, M.R.; Amini, E.; Kourosh-Arami, M.; Momeni, S.A.; Bokaie, S.; Sharifi, L. The Potential of T Cell Immunoglobulin and Mucin-Domain Containing-3 (Tim-3) in Designing Novel Immunotherapy for Bladder Cancer. Endocrine Metab. Immune Disord. Drug Targets 2021, 21, 2131–2146. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Toor, S.M.; Khalaf, S.; Elkord, E. Breast Cancer Cells and PD-1/PD-L1 Blockade Upregulate the Expression of PD-1, CTLA-4, TIM-3 and LAG-3 Immune Checkpoints in CD4+ T Cells. Vaccines 2019, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Fojnica, A.; Ljuca, K.; Akhtar, S.; Gatalica, Z.; Vranic, S. An Updated Review of the Biomarkers of Response to Immune Checkpoint Inhibitors in Merkel Cell Carcinoma: Merkel Cell Carcinoma and Immunotherapy. Cancers 2023, 15, 5084. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Hernández, A.; González Del Portillo, E.; Tamayo-Velasco, Á.; Figuero-Pérez, L.; Zhilina-Zhilina, S.; Fonseca-Sánchez, E.; Miramontes-González, J.P. Immune checkpoint inhibitors in non-small cell lung cancer: From current perspectives to future treatments—A systematic review. Ann. Transl. Med. 2023, 11, 354. [Google Scholar] [CrossRef]
- Fitzpatrick, O.; Naidoo, J. Immunotherapy for Stage III NSCLC: Durvalumab and Beyond. Lung Cancer Targets Ther. 2021, 12, 123–131. [Google Scholar] [CrossRef]
- Zou, Y.; Ren, X.; Zhang, H.; Wang, Y.; Wang, H.; Bai, R.; Zhang, Z.; Sun, G.; Xu, L. Efficacy and safety of durvalumab + chemotherapy vs. atezolizumab + chemotherapy in the treatment of small-cell lung cancer: A retrospective comparative cohort study. J. Thorac. Dis. 2023, 15, 3339–3349. [Google Scholar] [CrossRef]
- Semenescu, L.E.; Kamel, A.; Ciubotaru, V.; Baez-Rodriguez, S.M.; Furtos, M.; Costachi, A.; Dricu, A.; Tătăranu, L.G. An Overview of Systemic Targeted Therapy in Renal Cell Carcinoma, with a Focus on Metastatic Renal Cell Carcinoma and Brain Metastases. Curr. Issues Mol. Biol. 2023, 45, 7680–7704. [Google Scholar] [CrossRef]
- Tovoli, F.; De Lorenzo, S.; Trevisani, F. Immunotherapy with Checkpoint Inhibitors for Hepatocellular Carcinoma: Where Are We Now? Vaccines 2020, 8, 578. [Google Scholar] [CrossRef]
- Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2021, 70, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Lizardo, D.Y.; Kuang, C.; Hao, S.; Yu, J.; Huang, Y.; Zhang, L. Immunotherapy efficacy on mismatch repair-deficient colorectal cancer: From bench to bedside. Biochim. Biophys. Acta (BBA) Rev. Cancer 2020, 1874, 188447. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.D.; Mathias-Machado, M.C.; Jácome, A.; Gil, M.; Fogacci, J.; Sodré, B.; Passarini, T.; Chaves, A.; Diniz, P.H.; Lino, F.; et al. PD-L1 testing in advanced gastric cancer—What physicians who treat this disease must know—A literature review. J. Gastrointest. Oncol. 2023, 14, 1560–1575. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Liu, J.; Xu, K.; Chen, H.; Bian, C. PD-1/PD-L1 inhibitors for advanced or metastatic cervical cancer: From bench to bed. Front. Oncol. 2022, 12, 849352. [Google Scholar] [CrossRef]
- Corr, B.; Cosgrove, C.; Spinosa, D.; Guntupalli, S. Endometrial cancer: Molecular classification and future treatments. BMJ Med. 2022, 1, e000152. [Google Scholar] [CrossRef]
- Lin, A.Y.; Schnitter, J.M.; Gordon, L.I. I. Immune Checkpoint Blockade for the Treatment of Hodgkin Lymphoma. ImmunoTargets Ther. 2022, 11, 1–10. [Google Scholar] [CrossRef]
- Armand, P.; Rodig, S.; Melnichenko, V.; Thieblemont, C.; Bouabdallah, K.; Tumyan, G.; Özcan, M.; Portino, S.; Fogliatto, L.; Caballero, M.D.; et al. Pembrolizumab in Relapsed or Refractory Primary Mediastinal Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 3291–3299. [Google Scholar] [CrossRef] [PubMed]
- Franklin, M.R.; Platero, S.; Saini, K.S.; Curigliano, G.; Anderson, S. Immuno-oncology trends: Preclinical models, biomarkers, and clinical development. J. Immunother. Cancer 2022, 10, e003231. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Wu, H.; Wu, J.; Ding, P.; He, J.; Sang, M.; Liu, L. Mechanisms of immune checkpoint inhibitors: Insights into the regulation of circular RNAS involved in cancer hallmarks. Cell Death Dis. 2024, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Naimi, A.; Mohammed, R.N.; Raji, A.; Chupradit, S.; Yumashev, A.V.; Suksatan, W.; Shalaby, M.N.; Thangavelu, L.; Kamrava, S.; Shomali, N.; et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun. Signal. 2022, 20, 44. [Google Scholar] [CrossRef]
- Qiu, J.; Cheng, Z.; Jiang, Z.; Gan, L.; Zhang, Z.; Xie, Z. Immunomodulatory Precision: A Narrative Review Exploring the Critical Role of Immune Checkpoint Inhibitors in Cancer Treatment. Int. J. Mol. Sci. 2024, 25, 5490. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Mahmud, A.R.; Siddiquee, M.F.-.R.-S.; Shahriar, A.; Biswas, P.; Shimul, E.K.; Ahmed, S.Z.; Ema, T.I.; Rahman, N.; Khan, A.; et al. Role of T cells in cancer immunotherapy: Opportunities and challenges. Cancer Pathog. Ther. 2023, 1, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Khatwani, N.; Searles, T.G.; Turk, M.J.; Angeles, C.V. Memory CD8+ T cell responses to cancer. Semin. Immunol. 2020, 49, 101435. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Luo, S.; Ye, P.; Hwang, W.-L.; Cha, J.-H.; Yan, X.; Yang, W.-H. T-cell exhaustion and stemness in antitumor immunity: Characteristics, mechanisms, and implications. Front. Immunol. 2023, 14, 1104771. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ahn, E.; Kissick, H.T.; Ahmed, R. Reinvigorating Exhausted T Cells by Blockade of the PD-1 Pathway. Forum Immunopathol. Dis. Ther. 2015, 6, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Tie, Y.; Tang, F.; Wei, Y.-Q.; Wei, X.-W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 72. [Google Scholar] [CrossRef]
- Wang, M.M.; Coupland, S.E.; Aittokallio, T.; Figueiredo, C.R. Resistance to immune checkpoint therapies by tumour-induced T-cell desertification and exclusion: Key mechanisms, prognostication and new therapeutic opportunities. Br. J. Cancer 2023, 129, 1212–1224. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Yang, F.; Huang, Z.-Q.; Li, Y.-Y.; Shi, H.-Y.; Sun, Q.; Ma, Y.; Wang, Y.; Zhang, Y.; Yang, S.; et al. T cells, NK cells, and tumor-associated macrophages in cancer immunotherapy and the current state of the art of drug delivery systems. Front. Immunol. 2023, 14, 1199173. [Google Scholar] [CrossRef]
- Olbryt, M.; Rajczykowski, M.; Widłak, W. Biological Factors behind Melanoma Response to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 4071. [Google Scholar] [CrossRef]
- Khosravi, G.; Mostafavi, S.; Bastan, S.; Ebrahimi, N.; Gharibvand, R.S.; Eskandari, N. Immunologic tumor microenvironment modulators for turning cold tumors hot. Cancer Commun. 2024, 44, 521–553. [Google Scholar] [CrossRef] [PubMed]
- Kallingal, A.; Olszewski, M.; Maciejewska, N.; Brankiewicz, W.; Baginski, M. Cancer immune escape: The role of antigen presentation machinery. J. Cancer Res. Clin. Oncol. 2023, 149, 8131–8141. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, J.; Das, B.; Shin, S.; Chen, A. Challenges and Future Directions in the Management of Tumor Mutational Burden-High (TMB-H) Advanced Solid Malignancies. Cancers 2023, 15, 5841. [Google Scholar] [CrossRef]
- Yu, G.; Pang, Y.; Merchant, M.; Kesserwan, C.; Gangalapudi, V.; Abdelmaksoud, A.; Ranjan, A.; Kim, O.; Wei, J.S.; Chou, H.-C.; et al. Tumor Mutation Burden, Expressed Neoantigens and the Immune Microenvironment in Diffuse Gliomas. Cancers 2021, 13, 6092. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, T.; Jiang, C. Recent Advancements in Nanomedicine for ‘Cold’ Tumor Immunotherapy. Nano-Micro Lett. 2021, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Theocharopoulos, C.; Lialios, P.-P.; Foteinou, D.; Koumprentziotis, I.-A.; Xynos, G.; Gogas, H. Beyond CTLA-4 and PD-1 Inhibition: Novel Immune Checkpoint Molecules for Melanoma Treatment. Cancers 2023, 15, 2718. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Molina, A.M.; Santana, T.T.; Redondo, M.; Romero, M.J.B. The Crucial Role of Inflammation and the Immune System in Colorectal Cancer Carcinogenesis: A Comprehensive Perspective. Int. J. Mol. Sci. 2024, 25, 6188. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Huang, Y.Y.; Hurlstone, A. The role of IFN-γ-signalling in response to immune checkpoint blockade therapy. Essays Biochem. 2023, 67, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, R.; Martin, A.; Bommarito, D.; Wang, K.; Hansen, S.H.; Freeman, G.J.; Ritz, J. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. OncoImmunology 2015, 4, e1008824. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Lang, F.; Schrörs, B.; Löwer, M.; Türeci, Ö.; Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022, 21, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef] [PubMed]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef] [PubMed]
- Yin, N.; Li, X.; Zhang, X.; Xue, S.; Cao, Y.; Niedermann, G.; Lu, Y.; Xue, J. Development of pharmacological immunoregulatory anti-cancer therapeutics: Current mechanistic studies and clinical opportunities. Signal Transduct. Target. Ther. 2024, 9, 126. [Google Scholar] [CrossRef]
- Reiss, K.A.; Forde, P.M.; Brahmer, J.R. Harnessing the Power of the Immune System Via Blockade of PD-1 and PD-L1: A Promising New Anticancer Strategy. Immunotherapy 2014, 6, 459–475. [Google Scholar] [CrossRef]
- Cui, J.-W.; Li, Y.; Yang, Y.; Yang, H.-K.; Dong, J.-M.; Xiao, Z.-H.; He, X.; Guo, J.-H.; Wang, R.-Q.; Dai, B.; et al. Tumor immunotherapy resistance: Revealing the mechanism of PD-1/PD-L1-mediated tumor immune escape. Biomed. Pharmacother. 2024, 171, 116203. [Google Scholar] [CrossRef]
- Brunell, A.E.; Lahesmaa, R.; Autio, A.; Thotakura, A.K. Exhausted T cells hijacking the cancer-immunity cycle: Assets and liabilities. Front. Immunol. 2023, 14, 1151632. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Zamora, A.E.; Thomas, P.G.; Youngblood, B.A. Cell-Intrinsic Barriers of T Cell-Based Immunotherapy. Trends Mol. Med. 2016, 22, 1000–1011. [Google Scholar] [CrossRef]
- Wu, Q.; Yu, X.; Li, J.; Sun, S.; Tu, Y. Metabolic regulation in the immune response to cancer. Cancer Commun. 2021, 41, 661–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, W.; Gao, Y.; Zhang, Y.; Zhao, Y.; Hao, S.; Ni, T. The Role of Tumor Metabolic Reprogramming in Tumor Immunity. Int. J. Mol. Sci. 2023, 24, 17422. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.M.; George, S. Emerging resistance vs. losing response to immune check point inhibitors in renal cell carcinoma: Two differing phenomena. Cancer Drug Resist. 2023, 6, 642–655. [Google Scholar] [CrossRef]
- Salemme, V.; Centonze, G.; Avalle, L.; Natalini, D.; Piccolantonio, A.; Arina, P.; Morellato, A.; Ala, U.; Taverna, D.; Turco, E.; et al. The role of tumor microenvironment in drug resistance: Emerging technologies to unravel breast cancer heterogeneity. Front. Oncol. 2023, 13, 1170264. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Luo, Y.; Rao, D.; Wang, T.; Lei, Z.; Chen, X.; Zhang, B.; Li, Y.; Liu, B.; Xia, L.; et al. Myeloid-derived suppressor cells in cancer: Therapeutic targets to overcome tumor immune evasion. Exp. Hematol. Oncol. 2024, 13, 39. [Google Scholar] [CrossRef]
- Khouzam, R.A.; Brodaczewska, K.; Filipiak, A.; Zeinelabdin, N.A.; Buart, S.; Szczylik, C.; Kieda, C.; Chouaib, S. Tumor Hypoxia Regulates Immune Escape/Invasion: Influence on Angiogenesis and Potential Impact of Hypoxic Biomarkers on Cancer Therapies. Front. Immunol. 2021, 11, 613114. [Google Scholar] [CrossRef]
- Zhang, Y.; Brekken, R.A. Direct and indirect regulation of the tumor immune microenvironment by VEGF. J. Leukoc. Biol. 2022, 111, 1269–1286. [Google Scholar] [CrossRef]
- Moeckel, C.; Bakhl, K.; Georgakopoulos-Soares, I.; Zaravinos, A. The Efficacy of Tumor Mutation Burden as a Biomarker of Response to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2023, 24, 6710. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Escudero, M.; Arias-González, N.; Martínez-Cáceres, E. Regulatory cells and the effect of cancer immunotherapy. Mol. Cancer 2023, 22, 26. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jin, J.; Guo, D.; Tao, Z.; Hu, X. Immune Checkpoint Inhibitors Combined with Targeted Therapy: The Recent Advances and Future Potentials. Cancers 2023, 15, 2858. [Google Scholar] [CrossRef]
- Chyuan, I.-T.; Chu, C.-L.; Hsu, P.-N. Targeting the Tumor Microenvironment for Improving Therapeutic Effectiveness in Cancer Immunotherapy: Focusing on Immune Checkpoint Inhibitors and Combination Therapies. Cancers 2021, 13, 1188. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Li, Z.Z.; Zhong, N.N.; Cao, L.M.; Liu, B.; Bu, L.L. Charting new frontiers: Co-inhibitory immune checkpoint proteins in therapeutics, biomarkers, and drug delivery systems in cancer care. Transl. Oncol. 2023, 38, 101794. [Google Scholar] [CrossRef]
- Lao, Y.; Shen, D.; Zhang, W.; He, R.; Jiang, M. Immune Checkpoint Inhibitors in Cancer Therapy—How to Overcome Drug Resistance? Cancers 2022, 14, 3575. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Kang, K.; Chen, P.; Zeng, Z.; Li, G.; Xiong, W.; Yi, M.; Xiang, B. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol. Cancer 2024, 23, 108. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hsu, J.-M.; Sun, L.; Wang, S.-C.; Hung, M.-C. Advances and prospects of biomarkers for immune checkpoint inhibitors. Cell Rep. Med. 2024, 5, 101621. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Jalal, S.; Earley, J.N.; Turchi, J.J. DNA repair: From genome maintenance to biomarker and therapeutic target. Clin. Cancer Res. 2011, 17, 6973–6984. [Google Scholar] [CrossRef]
- Chae, Y.K.; Anker, J.F.; Bais, P.; Namburi, S.; Giles, F.J.; Chuang, J.H. Mutations in DNA repair genes are associated with increased neo-antigen load and activated T cell infiltration in lung adenocarcinoma. Oncotarget 2018, 9, 7949–7960. [Google Scholar] [CrossRef] [PubMed]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from pd-1/pd-l1 blockade in advanced urothelial cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Amrane, K.; Le Meur, C.; Besse, B.; Hemon, P.; Le Noac’h, P.; Pradier, O.; Berthou, C.; Abgral, R.; Uguen, A. HLA-DR expression in melanoma: From misleading therapeutic target to potential immunotherapy biomarker. Front. Immunol. 2023, 14, 1285895. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.Y.; Zhao, L.; Green, M.D.; Ravishankar, S.; Towlerton, A.M.H.; Scott, A.J.; Raghavan, M.; Cusick, M.F.; Warren, E.H.; Ramnath, N. Class II HLA-DRB4 is a predictive biomarker for survival following immunotherapy in metastatic non-small cell lung cancer. Sci. Rep. 2024, 14, 345. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, B.; Bieche, I.; Pasmant, E.; Hamzaoui, N.; Leulliot, N.; Michon, L.; de Reynies, A.; Attignon, V.; Foote, M.B.; Masliah-Planchon, J.; et al. PD-1 Blockade in Solid Tumors with Defects in Polymerase Epsilon. Cancer Discov. 2022, 12, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Corgnac, S.; Malenica, I.; Mezquita, L.; Auclin, E.; Voilin, E.; Kacher, J.; Halse, H.; Grynszpan, L.; Signolle, N.; Dayris, T.; et al. CD103+CD8+ TRM Cells Accumulate in Tumors of Anti-PD-1-Responder Lung Cancer Patients and Are Tumor-Reactive Lymphocytes Enriched with Tc17. Cell Rep. Med. 2020, 1, 100127. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, J.Y.; Jeon, S.H.; Nam, H.; Jung, J.H.; Jeon, M.; Kim, E.-S.; Bae, S.J.; Ahn, J.; Yoo, T.-K.; et al. CD39 + tissue-resident memory CD8 + T cells with a clonal overlap across compartments mediate antitumor immunity in breast cancer. Sci. Immunol. 2022, 7, eabn8390. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hu, X.; Feng, K.; Gao, R.; Xue, Z.; Zhang, S.; Zhang, Y.; Corse, E.; Hu, Y.; Han, W.; et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 2020, 3, 108–121. [Google Scholar] [CrossRef]
- Chen, X.; Su, C.; Ren, S.; Zhou, C.; Jiang, T. Pan-cancer analysis of KEAP1 mutations as biomarkers for immunotherapy outcomes. Ann. Transl. Med. 2020, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Papillon-Cavanagh, S.; Doshi, P.; Dobrin, R.; Szustakowski, J.; Walsh, A.M. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open 2022, 5, e000706. [Google Scholar] [CrossRef]
- Di Federico, A.; De Giglio, A.; Parisi, C.; Gelsomino, F. STK11/LKB1 and KEAP1 mutations in non-small cell lung cancer: Prognostic rather than predictive? Eur. J. Cancer 2021, 157, 108–113. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, M.; Ciappina, G.; Spagnolo, C.C.; Squeri, A.; Passalacqua, M.I.; Aguilar, A.; Gonzalez-Cao, M.; Giovannetti, E.; Silvestris, N.; Rosell, R. Targeted therapies for KRAS-mutant non-small cell lung cancer: From preclinical studies to clinical development—A narrative review. Transl. Lung Cancer Res. 2023, 12, 346–368. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1–NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Sivakumar, S.; Schrock, A.B.; Madison, R.; Fabrizio, D.; Gjoerup, O.; Ross, J.S.; Frampton, G.M.; Napalkov, P.; Montesion, M.; et al. Comprehensive pan-cancer genomic landscape of KRAS altered cancers and real-world outcomes in solid tumors. npj Precis. Oncol. 2022, 6, 91. [Google Scholar] [CrossRef]
- Vranic, S.; Gatalica, Z. PD-L1 testing by immunohistochemistry in Immuno-Oncology. Bosn. J. Basic Med. Sci. 2022, 23, 15–25. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, Z.; Zhang, W.; Zhang, W.; Buzdin, A.; Mu, X.; Yan, Q.; Zhao, X.; Chang, H.-H.; Duhon, M.; et al. FDA-Approved and emerging next generation predictive biomarkers for immune checkpoint inhibitors in cancer patients. Front. Oncol. 2021, 11, 683419. [Google Scholar] [CrossRef] [PubMed]
- Abad, M.N.; Calabuig-Fariñas, S.; de Mena, M.L.; Torres-Martínez, S.; González, C.G.; García, J.G.; González-Cruz, V.I.; Herrero, C.C. Programmed death-ligand 1 (PD-L1) as immunotherapy biomarker in breast cancer. Cancers 2022, 14, 307. [Google Scholar] [CrossRef]
- Li, J.-X.; Huang, J.-M.; Jiang, Z.-B.; Li, R.-Z.; Sun, A.; Leung, E.L.-H.; Yan, P.-Y. Current clinical progress of PD-1/PD-L1 immunotherapy and potential combination treatment in non–small cell lung cancer. Integr. Cancer Ther. 2019, 18, 1534735419890020. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human t cell stimulation, but Only receptor ligation prevents t cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Schwartz, J.C.; Guo, X.; Bhatia, S.; Cao, E.; Lorenz, M.; Cammer, M.; Chen, L.; Zhang, Z.Y.; Edidin, M.A.; et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 2004, 20, 337–347. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Wang, J.; Yoshida, T.; Nakaki, F.; Hiai, H.; Okazaki, T.; Honjo, T. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. USA 2005, 102, 11823–11828. [Google Scholar] [CrossRef]
- Wang, J.; Okazaki, I.-M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell–mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Agata, Y.; Kawasaki, A.; Sato, M.; Imamura, S.; Minato, N.; Yagita, H.; Nakano, T.; Honjo, T. Developmentally regulated expression of the PD-1 protein on the surface of double-negative(CD4−CD8−) thymocytes. Int. Immunol. 1996, 8, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of treg cells, and enhanced expression of the PD-1 costimulatory pathway. J. Neurosci. Res. 2006, 84, 370–378. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical Signaling of PD-1 on T Cells and Its Functional Implications. Cancer J. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Cho, H.Y.; Lee, S.W.; Seo, S.K.; Choi, I.W.; Choi, I.; Lee, S.W. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-α-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochim. Biophys. Acta 2008, 1779, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2016, 8, 2171–2186. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed Death-1 Ligand 1 Interacts Specifically with the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef]
- Mueller, S.N.; Vanguri, V.K.; Ha, S.-J.; West, E.E.; Keir, M.E.; Glickman, J.N.; Sharpe, A.H.; Ahmed, R. PD-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice. J. Clin. Investig. 2010, 120, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Akiba, H.; Koyanagi, A.; Azuma, M.; Yagita, H.; Okumura, K. Blockade of B7-H1 on Macrophages Suppresses CD4+ T Cell Proliferation by Augmenting IFN-γ-Induced Nitric Oxide Production. J. Immunol. 2005, 175, 1586–1592. [Google Scholar] [CrossRef]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef]
- Probst, H.C.; McCoy, K.; Okazaki, T.; Honjo, T.; van den Broek, M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol. 2005, 6, 280–286. [Google Scholar] [CrossRef]
- Nava, S.; Lisini, D.; Frigerio, S.; Bersano, A. Dendritic Cells and Cancer Immunotherapy: The Adjuvant Effect. Int. J. Mol. Sci. 2021, 22, 12339. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.-H.; Chan, L.-C.; Li, C.-W.; Hsu, J.L.; Hung, M.-C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Schöniger, S.; Jasani, B. The PD-1/PD-L1 pathway: A perspective on comparative immuno-oncology. Animals 2022, 12, 2661. [Google Scholar] [CrossRef]
- Zhao, Y.; Harrison, D.L.; Song, Y.; Ji, J.; Huang, J.; Hui, E. Antigen-Presenting Cell-Intrinsic PD-1 Neutralizes PD-L1 in cis to Attenuate PD-1 Signaling in T Cells. Cell Rep. 2018, 24, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Viguera, E.; Canceill, D.; Ehrlich, S. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 2001, 20, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Angelis, G.L.D.; Bottarelli, L.; Azzoni, C.; Angelis, N.D.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite instability in colorectal cancer. Acta Biomed. 2018, 89, 97–101. [Google Scholar] [CrossRef]
- Weissenbach, J.; Gyapay, G.; Dib, C.; Vignal, A.; Morissette, J.; Millasseau, P.; Vaysseix, G.; Lathrop, M. A second-generation linkage map of the human genome. Nature 1992, 359, 794–801. [Google Scholar] [CrossRef]
- Arzimanoglou, I.I.; Gilbert, F.; Barber, H.R. Microsatellite instability in human solid tumors. Cancer 1998, 82, 1808–1820. [Google Scholar] [CrossRef]
- Gatalica, Z.; Vranic, S.; Xiu, J.; Swensen, J.; Reddy, S. High microsatellite instability (MSI-H) colorectal carcinoma: A brief review of predictive biomarkers in the era of personalized medicine. Fam. Cancer 2016, 15, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. Implications of mismatch repair-deficient status on management of early stage colorectal cancer. J. Gastrointest. Oncol. 2015, 6, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer—The stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e2073. [Google Scholar] [CrossRef] [PubMed]
- Jo, W.-S.; Carethers, J.M. Chemotherapeutic implications in microsatellite unstable colorectal cancer1. Cancer Biomarkers 2006, 2, 51–60. [Google Scholar] [CrossRef]
- Profir, M.; Roşu, O.A.; Creţoiu, S.M.; Gaspar, B.S. Friend or foe: Exploring the relationship between the gut microbiota and the pathogenesis and treatment of digestive cancers. Microorganisms 2024, 12, 955. [Google Scholar] [CrossRef]
- Imai, K.; Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 2007, 29, 673–680. [Google Scholar] [CrossRef]
- Plazzer, J.P.; Sijmons, R.H.; Woods, M.O.; Peltomäki, P.; Thompson, B.; Dunnen, J.T.D.; Macrae, F. The InSiGHT database: Utilizing 100 years of insights into lynch syndrome. Fam. Cancer 2013, 12, 175–180. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J.; et al. Screening for the lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef]
- Chen, S.; Wang, W.; Lee, S.; Nafa, K.; Lee, J.; Romans, K.; Watson, P.; Gruber, S.B.; Euhus, D.; Kinzler, K.W.; et al. Prediction of germline mutations and cancer risk in the lynch syndrome. JAMA 2006, 296, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.; Mukherjee, B.; Raymond, V.M.; Tayob, N.; Kastrinos, F.; Sparr, J.; Wang, F.; Bandipalliam, P.; Syngal, S.; Gruber, S.B. Calculation of risk of colorectal and endometrial cancer among patients with lynch syndrome. Gastroenterology 2009, 137, 1621–1627. [Google Scholar] [CrossRef]
- Roth, A.D.; Tejpar, S.; Yan, P.; Fiocca, R.; Dietrich, D.; Delorenzi, M.; Labianca, R.; Cunningham, D.; Van Cutsem, E.; Bosman, F. Stage-specific prognostic value of molecular markers in colon cancer: Results of the translational study on the PETACC 3-EORTC 40993-SAKK 60–00 trial. J. Clin. Oncol. 2009, 27, 4002. [Google Scholar] [CrossRef]
- Koopman, M.; Kortman, G.A.M.; Mekenkamp, L.; Ligtenberg, M.J.L.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.A.; van Krieken, J.H.J.M. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Smoot, D.T.; Carethers, J.M.; Rahmanian, M.; Kittles, R.; Vosganian, G.; Doura, M.; Nidhiry, E.; Naab, T.; Momen, B.; et al. High incidence of microsatellite instability in colorectal cancer from African Americans. Clin. Cancer Res. 2003, 9, 1112–1117. [Google Scholar] [PubMed]
- Kumar, K.; Brim, H.; Giardiello, F.; Smoot, D.T.; Nouraie, M.; Lee, E.L.; Ashktorab, H. Distinct BRAF (V600E) and KRAS mutations in high microsatellite instability sporadic colorectal cancer in african americans. Clin. Cancer Res. 2009, 15, 1155–1161. [Google Scholar] [CrossRef]
- Soliman, A.S.; Bondy, M.L.; El-Badawy, S.A.; Mokhtar, N.; Eissa, S.; Bayoumy, S.; Seifeldin, I.A.; Houlihan, P.S.; Lukish, J.R.; Watanabe, T.; et al. Contrasting molecular pathology of colorectal carcinoma in Egyptian and Western patients. Br. J. Cancer 2001, 85, 1037–1046. [Google Scholar] [CrossRef]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef]
- Lawes, D.; SenGupta, S.; Boulos, P. The clinical importance and prognostic implications of microsatellite instability in sporadic cancer. Eur. J. Surg. Oncol. (EJSO) 2003, 29, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Poynter, J.N.; Siegmund, K.D.; Weisenberger, D.J.; Long, T.I.; Thibodeau, S.N.; Lindor, N.; Young, J.; Jenkins, M.A.; Hopper, J.L.; Baron, J.A.; et al. Molecular characterization of msi-h colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 3208–3215. [Google Scholar] [CrossRef]
- Funkhouser, W.K.; Lubin, I.M.; Monzon, F.A.; Zehnbauer, B.A.; Evans, J.P.; Ogino, S.; Nowak, J.A. Relevance, pathogenesis, and testing algorithm for mismatch repair–defective colorectal carcinomas: A report of the association for molecular pathology. J. Mol. Diagn. 2012, 14, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Watanabe, T.; Wu, T.-T.; Rashid, A.; Li, S.; Hamilton, S.R. Histopathological identification of colon cancer with microsatellite instability. Am. J. Pathol. 2001, 158, 527–535. [Google Scholar] [CrossRef]
- Greenson, J.K.; Bonner, J.D.; Ben-Yzhak, O.; Cohen, H.I.; Miselevich, I.; Resnick, M.B.; Trougouboff, P.; Tomsho, L.D.; Kim, E.; Low, M.; et al. Phenotype of microsatellite unstable colorectal carcinomas: Well-differentiated and focally mucinous tumors and the absence of dirty necrosis correlate with microsatellite instability. Am. J. Surg. Pathol. 2003, 27, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Shia, J.; Ellis, N.A.; Paty, P.B.; Nash, G.M.; Qin, J.; Offit, K.; Zhang, X.-M.; Markowitz, A.J.; Nafa, K.; Guillem, J.G.; et al. Value of histopathology in predicting microsatellite instability in hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer. Am. J. Surg. Pathol. 2003, 27, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Yearsley, M.; Hampel, H.; Lehman, A.; Nakagawa, H.; de la Chapelle, A.; Frankel, W.L. Histologic features distinguish microsatellite-high from microsatellite-low and microsatellite-stable colorectal carcinomas, but do not differentiate germline mutations from methylation of the MLH1 promoter. Hum. Pathol. 2006, 37, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Greenson, J.K.; Huang, S.-C.; Herron, C.B.; Moreno, V.; Bonner, J.D.; Tomsho, L.P.B.; Ben-Izhak, O.; Cohen, H.I.; Trougouboff, P.; Bejhar, J.; et al. Pathologic predictors of microsatellite instability in colorectal cancer. Am. J. Surg. Pathol. 2009, 33, 126–133. [Google Scholar] [CrossRef]
- Zlobec, I.; Bihl, M.P.; Foerster, A.; Rufle, A.; Lugli, A. The impact of CpG island methylator phenotype and microsatellite instability on tumour budding in colorectal cancer. Histopathology 2012, 61, 777–787. [Google Scholar] [CrossRef]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef]
- Said, S.S.; Ibrahim, W.N. Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives. Pharmaceutics 2023, 15, 1143. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. J. Clin. Investig. 2019, 4, 126908. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden–High Solid Tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Rousseau, B.; Foote, M.B.; Maron, S.B.; Diplas, B.H.; Lu, S.; Argilés, G.; Cercek, A.; Diaz, L.A. The Spectrum of Benefit from Checkpoint Blockade in Hypermutated Tumors. New Engl. J. Med. 2021, 384, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Gurjao, C.; Tsukrov, D.; Imakaev, M.; Luquette, L.; Mirny, L. Limited evidence of tumour mutational burden as a biomarker of response to immunotherapy. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ionescu, R.F.; Cozma, E.C.; Enache, R.M.; Cretoiu, S.M.; Iancu, M.A.; Mandea, M.; Profir, M.; Roşu, O.A.; Gaspar, B.S. Intestinal Microbiomics in Physiological and Pathological Conditions. In Advances in Probiotics for Health and Nutrition; Vasudeo, Z., Mohd, F.M.D., Puja, G., Bhupendra Gopalbhai, P., Eds.; IntechOpen: Rijeka, Croatia, 2023. [Google Scholar] [CrossRef]
- Yi, M.; Qin, S.; Chu, Q.; Wu, K. The role of gut microbiota in immune checkpoint inhibitor therapy. HepatoBiliary Surg. Nutr. 2018, 7, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lv, J.; Guo, F.; Li, J.; Jia, Y.; Jiang, D.; Wang, N.; Zhang, C.; Kong, L.; Liu, Y.; et al. Gut Microbiome Influences the Efficacy of PD-1 Antibody Immunotherapy on MSS-Type Colorectal Cancer via Metabolic Pathway. Front. Microbiol. 2020, 11, 814. [Google Scholar] [CrossRef] [PubMed]
- Naqash, A.R.; Kihn-Alarcón, A.J.; Stavraka, C.; Kerrigan, K.; Vareki, S.M.; Pinato, D.J.; Puri, S. The role of gut microbiome in modulating response to immune checkpoint inhibitor therapy in cancer. Ann. Transl. Med. 2021, 9, 1034. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; de Vos, W.M.; Montijn, R.C.; Roeselers, G. Differential Modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio 2014, 5, e01438-14. [Google Scholar] [CrossRef]
- Ottman, N.; Huuskonen, L.; Reunanen, J.; Boeren, S.; Klievink, J.; Smidt, H.; Belzer, C.; de Vos, W.M. Characterization of Outer Membrane Proteome of Akkermansia muciniphila Reveals Sets of Novel Proteins Exposed to the Human Intestine. Front. Microbiol. 2016, 7, 1157. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, R.F.; Enache, R.M.; Cretoiu, S.M.; Gaspar, B.S. Gut Microbiome Changes in Gestational Diabetes. Int. J. Mol. Sci. 2022, 23, 12839. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Clavel, T.; Gomes-Neto, J.C.; Lagkouvardos, I.; Ramer-Tait, A.E. Deciphering interactions between the gut microbiota and the immune system via microbial cultivation and minimal microbiomes. Immunol. Rev. 2017, 279, 8–22. [Google Scholar] [CrossRef]
- Everts, B. Metabolomics in Immunology Research. Methods Mol. Biol. 2018, 1730, 29–42. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Younis, A.; Gribben, J. Immune Checkpoint Inhibitors: Fundamental Mechanisms, Current Status and Future Directions. Immuno 2024, 4, 186–210. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Choi, J.; Lee, S.Y. Clinical Characteristics and Treatment of Immune-Related Adverse Events of Immune Checkpoint Inhibitors. Immune Netw. 2020, 20, e9. [Google Scholar] [CrossRef] [PubMed]
- Spiers, L.; Coupe, N.; Payne, M. Toxicities associated with checkpoint inhibitors—An overview. Rheumatology 2019, 58, vii7–vii16. [Google Scholar] [CrossRef] [PubMed]
- Shalitin, S. Endocrine-related adverse conditions in pediatric patients treated with immune checkpoint inhibition for malignancies. Horm. Res. Paediatr. 2024, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ciurej, A.; Lewis, E.; Gupte, A.; Al-Antary, E. Checkpoint Immunotherapy in Pediatric Oncology: Will We Say Checkmate Soon? Vaccines 2023, 11, 1843. [Google Scholar] [CrossRef] [PubMed]
- Dagar, G.; Gupta, A.; Shankar, A.; Chauhan, R.; Macha, M.A.; Bhat, A.A.; Das, D.; Goyal, R.; Bhoriwal, S.; Pandita, R.K.; et al. The future of cancer treatment: Combining radiotherapy with immunotherapy. Front. Mol. Biosci. 2024, 11, 1409300. [Google Scholar] [CrossRef]
- Warszyńska, M.; Repetowski, P.; Dąbrowski, J.M. Photodynamic therapy combined with immunotherapy: Recent advances and future research directions. Coord. Chem. Rev. 2023, 495, 215350. [Google Scholar] [CrossRef]
- Profir, M.; Roşu, O.A.; Gaspar, B.S.; Cretoiu, S.M. Gut Microbiome and the Role of Its Metabolites as Promoters or Inhibitors in Gastrointestinal Cancers. In Interdisciplinary Cancer Research; Springer International Publishing: Cham, Switzerland, 2024; pp. 1–27. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Target Molecule | Checkpoint Inhibitor Name | Brand Name | First Approval FDA (USA) | First Approval EMA * (EU) | First Cancer Type Approved |
|---|---|---|---|---|---|
| PD-1 | Pembrolizumab [21] | Keytruda | September 2014 | July 2015 | Melanoma |
| Nivolumab [22] ** | Opdivo | December 2014 | June 2015 | Melanoma | |
| Cemiplimab [23] | Libtayo | September 2018 | June 2019 | Cutaneous squamous cell carcinoma (CSCC) | |
| Dostalimab [24] | Jemperli | April 2021 | April 2021 | Endometrial cancer with dMMR/MSI-H status | |
| Toripalimab [25] | Loqtorzi | October 2023 | ongoing | Nasopharyngeal carcinoma | |
| PD-L1 | Atezolizumab [26] | Tecentriq | May 2016 | September 2017 | UC |
| Durvalumab [27] | Imfinzi | May 2017 | September 2018 | NSCLC | |
| Avelumab [28] | Bavencio | March 2017 | September 2017 | Merkel cell carcinoma | |
| CTLA-4 | Ipilimumab [29] ** | Yervoy | March 2011 | July 2011 | Melanoma |
| Tremelimumab [30] | Imjudo | October 2022 | December 2022 | HCC (in combination) |
| Predictive Biomarker | Cancer Type | Drugs |
|---|---|---|
| KRAS G12C [117] | NSCLC, pancreatic ductal adenocarcinoma and CRC | Sotorasib, Adagrasib (approved) |
| Advanced or metastatic solid tumors | GDC-6036—monotherapy or in combination with Cetuximab, Atezolizumab, Bevacizumab | |
| Advanced solid tumors | JDQ443—monotherapy or in combination with TNO155 or Tislelizumab | |
| Still in phase I/II studies | D-1553, LY3537982, JAB-21822 | |
| KRAS G12D [117] | Still in preclinical studies | MRTX1133—long half-life, potency, and antitumor activity |
| KRAS G12D, G12V, G13D, and G12C [117] | Still in phase I study in patients with metastatic NSCLC, CRC, or pancreatic adenocarcinoma | V941—lipid nanoparticle-formulated mRNA-based cancer vaccine, potential immunostimulatory and antineoplastic activities |
| NRF2-KEAP1 axis [116] | Estrogen receptors (ERs) + metastatic breast cancer | SFX-01—safe and efficacious when used in combination with aromatase inhibitors, Tamoxifen and Fulvestrant |
| NRF2 inhibitors [118] | Still in studies—lymphocytic leukemia tumor cells and many types of solid cancer cells | Brusatol |
| Still in preclinical models—NSCLC cells with KEAP1 mutations | ML385—combined with Doxorubicin, Taxol, or Carboplatin | |
| Phase 2 clinical trials for cancer | Halofuginone—inhibitor of collagen type I synthesis and of prolyl-tRNA synthetase | |
| Phase 2 clinical trial for bladder cancer treatment | HT-100—oral analog of halofuginone |
| Favorable Response | Poor Response |
|---|---|
| Clostridiales [16] | Bacteroidales [16,198] |
| Faecalibacterium [16] | Escherichia coli [16,206] |
| Ruminococcaceae [16] | Anaerotruncus colihominis [16] |
| Prevotella [198] | Bacteroides [198] |
| Akkermansia muciniphila [198] | |
| Bifidobacterium [199] | |
| Lactobacillus [199] | |
| Klebsiella pneumoniae [206] | |
| Streptococcus thermophiles [206] | |
| Lachnospiraceae [206] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavelescu, L.A.; Enache, R.M.; Roşu, O.A.; Profir, M.; Creţoiu, S.M.; Gaspar, B.S. Predictive Biomarkers and Resistance Mechanisms of Checkpoint Inhibitors in Malignant Solid Tumors. Int. J. Mol. Sci. 2024, 25, 9659. https://doi.org/10.3390/ijms25179659
Pavelescu LA, Enache RM, Roşu OA, Profir M, Creţoiu SM, Gaspar BS. Predictive Biomarkers and Resistance Mechanisms of Checkpoint Inhibitors in Malignant Solid Tumors. International Journal of Molecular Sciences. 2024; 25(17):9659. https://doi.org/10.3390/ijms25179659
Chicago/Turabian StylePavelescu, Luciana Alexandra, Robert Mihai Enache, Oana Alexandra Roşu, Monica Profir, Sanda Maria Creţoiu, and Bogdan Severus Gaspar. 2024. "Predictive Biomarkers and Resistance Mechanisms of Checkpoint Inhibitors in Malignant Solid Tumors" International Journal of Molecular Sciences 25, no. 17: 9659. https://doi.org/10.3390/ijms25179659
APA StylePavelescu, L. A., Enache, R. M., Roşu, O. A., Profir, M., Creţoiu, S. M., & Gaspar, B. S. (2024). Predictive Biomarkers and Resistance Mechanisms of Checkpoint Inhibitors in Malignant Solid Tumors. International Journal of Molecular Sciences, 25(17), 9659. https://doi.org/10.3390/ijms25179659