
Pangenome Identification and Analysis of Terpene Synthase Gene Family Members in Gossypium
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. A Syntelog-Based Pangenome of Gossypium Genus
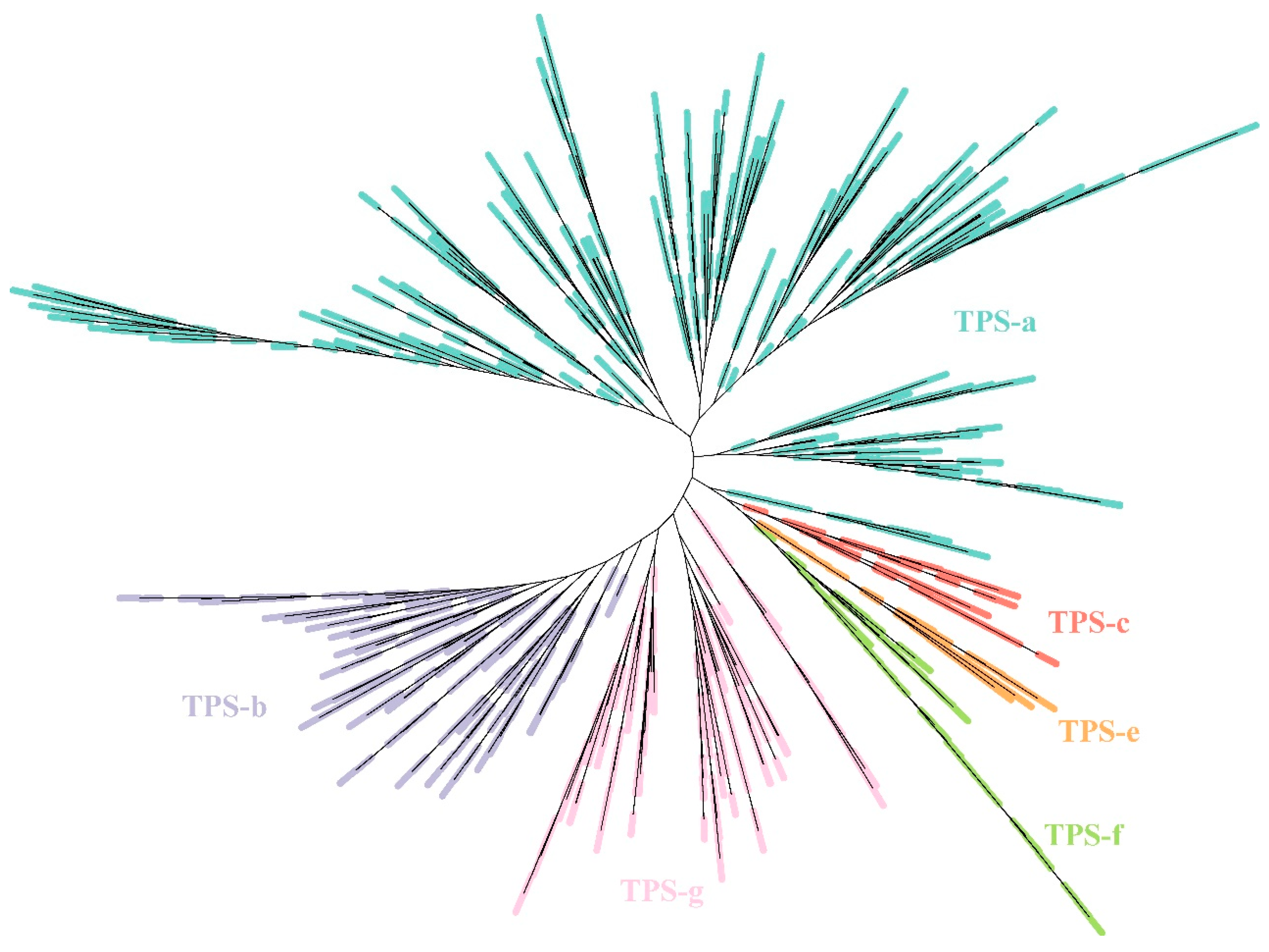
2.2. Identification of TPS Gene Family Based on Gossypium Pangenome
2.3. Mutational Load Analysis and Selection Pressure Analysis of Identified TPS Genes
2.4. Gene Structure Analysis of Representative TPS Genes across Cotton Species
2.5. Atypical TPS Genes in the Gossypium Pangenome
2.6. Identification of Herbivory-Responsive TPS Genes Using RNA-Seq Data
3. Discussion
4. Materials and Methods
4.1. Construction of a Syntelog-Based Pangenome for Gossypium Using Synteny
4.2. Identification of TPS Gene Family and Construction of the Phylogenetic Tree
4.3. The Mutational Load of TPS Genes in Different Gossypium Species
4.4. Ka/Ks Calculation
4.5. RNA-Seq Analysis
4.6. Conserved Motifs and Gene Structure Analysis
4.7. Coexpression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, X.Z.; Xiao, Y.T.; Köllner, T.G.; Jing, W.X.; Kou, J.F.; Chen, J.Y.; Liu, D.F.; Gu, S.H.; Wu, J.X.; Zhang, Y.J.; et al. The terpene synthase gene family in Gossypium hirsutum harbors a linalool synthase GhTPS12 implicated in direct defence responses against herbivores. Plant Cell Environ. 2018, 41, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Kutty, N.N.; Mishra, M. Dynamic distress calls: Volatile info chemicals induce and regulate defense responses during herbivory. Front. Plant Sci. 2023, 14, 1135000. [Google Scholar] [CrossRef] [PubMed]
- Turlings, T.C.; Erb, M. Tritrophic interactions mediated by herbivore-induced plant volatiles: Mechanisms, ecological relevance, and application potential. Annu. Rev. Entomol. 2018, 63, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Erb, M. Volatile uptake, transport, perception, and signaling shape a plant’s nose. Essays Biochem. 2022, 66, 695–702. [Google Scholar]
- Rosenkranz, M.; Chen, Y.; Zhu, P.; Vlot, A.C. Volatile terpenes–mediators of plant-to-plant communication. Plant J. 2021, 108, 617–631. [Google Scholar] [CrossRef]
- Irmisch, S.; Jiang, Y.; Chen, F.; Gershenzon, J.; Köllner, T.G. Terpene synthases and their contribution to herbivore-induced volatile emission in western balsam poplar (Populus trichocarpa). BMC Plant Biol. 2014, 14, 270. [Google Scholar] [CrossRef] [PubMed]
- Takabayashi, J. Herbivory-induced plant volatiles mediate multitrophic relationships in ecosystems. Plant Cell Physiol. 2022, 63, 1344–1355. [Google Scholar] [CrossRef]
- Zhou, F.; Pichersky, E. More is better: The diversity of terpene metabolism in plants. Curr. Opin. Plant Biol. 2020, 55, 1–10. [Google Scholar] [CrossRef]
- Cofer, T.M.; Seidl-Adams, I.; Tumlinson, J.H. From acetoin to (Z)-3-hexen-1-ol: The diversity of volatile organic compounds that induce plant responses. J. Agr. Food Chem. 2018, 66, 11197–11208. [Google Scholar] [CrossRef]
- Hu, L. Integration of multiple volatile cues into plant defense responses. New Phytol. 2022, 233, 618–623. [Google Scholar] [CrossRef]
- Mauch-Mani, B.; Baccelli, I.; Luna, E.; Flors, V. Defense priming: An adaptive part of induced resistance. Annu. Rev. Plant Biol. 2017, 68, 485–512. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Pichersky, E. The complete functional characterisation of the terpene synthase family in tomato. New Phytol. 2020, 226, 1341–1360. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Chen, X.; Wang, M.; Bach, T.J.; Chye, M.L. Improved fruit α-tocopherol, carotenoid, squalene and phytosterol contents through manipulation of Brassica juncea 3-HYDROXY-3-METHYLGLUTARYL-COA SYNTHASE 1 in transgenic tomato. Plant Biotechnol. J. 2018, 16, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef]
- Li, M.; Li, X.; Zhou, J.; Sun, Y.; Du, J.; Wang, Z.; Luo, Y.; Zhang, Y.; Chen, Q.; Wang, Y.; et al. Genome-wide identification and analysis of terpene synthase (TPS) genes in celery reveals their regulatory roles in terpenoid biosynthesis. Front. Plant Sci. 2022, 13, 1010780. [Google Scholar] [CrossRef]
- Wang, W.; Wang, M.Y.; Zeng, Y.; Chen, X.; Wang, X.; Barrington, A.M.; Tao, J.; Atkinson, R.G.; Nieuwenhuizen, N.J. The terpene synthase (TPS) gene family in kiwifruit shows high functional redundancy and a subset of TPS likely fulfil overlapping functions in fruit flavour, floral bouquet and defence. Mol. Hortic. 2023, 3, 9. [Google Scholar] [CrossRef]
- Aubourg, S.; Lecharny, A.; Bohlmann, J. Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana. Mol. Genet. Genom. 2002, 267, 730–745. [Google Scholar] [CrossRef]
- Chen, H.; Guo, M.; Dong, S.; Wu, X.; Zhang, G.; He, L.; Jiao, Y.; Chen, S.; Li, L.; Luo, H. A chromosome-scale genome assembly of Artemisia argyi reveals unbiased subgenome evolution and key contributions of gene duplication to volatile terpenoid diversity. Plant Commun. 2023, 4, 100516. [Google Scholar] [CrossRef]
- Liu, Z.; Fu, Y.; Wang, H.; Zhang, Y.; Han, J.; Wang, Y.; Shen, S.; Li, C.; Jiang, M.; Yang, X.; et al. The high-quality sequencing of the Brassica rapa ‘XiangQingCai’genome and exploration of genome evolution and genes related to volatile aroma. Hortic. Res. 2023, 10, uhad187. [Google Scholar] [CrossRef]
- Booth, J.K.; Yuen, M.M.; Jancsik, S.; Madilao, L.L.; Page, J.E.; Bohlmann, J. Terpene synthases and terpene variation in Cannabis sativa. Plant Physiol. 2020, 184, 130–147. [Google Scholar] [CrossRef]
- Yang, Z.; Zhan, T.; Xie, C.; Huang, S.; Zheng, X. Genome-wide analyzation and functional characterization on the TPS family provide insight into the biosynthesis of mono-terpenes in the camphor tree. Plant Physiol. Bioch. 2023, 196, 55–64. [Google Scholar] [CrossRef]
- Alquézar, B.; Rodríguez, A.; de la Peña, M.; Peña, L. Genomic analysis of terpene synthase family and functional characterization of seven sesquiterpene synthases from Citrus sinensis. Front. Plant Sci. 2017, 8, 1481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, G.Q.; Zhang, D.; Liu, X.D.; Xu, X.Y.; Sun, W.H.; Yu, X.; Zhu, X.; Wang, Z.W.; Zhao, X.; et al. Chromosome-scale assembly of the Dendrobium chrysotoxum genome enhances the understanding of orchid evolution. Hortic. Res. 2021, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Külheim, C.; Padovan, A.; Hefer, C.; Krause, S.T.; Köllner, T.G.; Myburg, A.A.; Degenhardt, J.; Foley, W.J. The Eucalyptus terpene synthase gene family. BMC Genom. 2015, 16, 450. [Google Scholar] [CrossRef]
- Bao, T.; Shadrack, K.; Yang, S.; Xue, X.; Li, S.; Wang, N.; Wang, Q.; Wang, L.; Gao, X.; Cronk, Q. Functional characterization of terpene synthases accounting for the volatilized-terpene heterogeneity in Lathyrus odoratus cultivar flowers. Plant Cell Physiol. 2020, 61, 1733–1749. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, N.J.; Green, S.A.; Chen, X.; Bailleul, E.J.; Matich, A.J.; Wang, M.Y.; Atkinson, R.G. Functional genomics reveals that a compact terpene synthase gene family can account for terpene volatile production in apple. Plant Physiol. 2013, 161, 787–804. [Google Scholar] [CrossRef]
- Han, X.; Zhang, J.; Han, S.; Chong, S.L.; Meng, G.; Song, M.; Wang, Y.; Zhou, S.; Liu, C.; Lou, L.; et al. The chromosome-scale genome of Phoebe bournei reveals contrasting fates of terpene synthase (TPS)-a and TPS-b subfamilies. Plant Commun. 2022, 3, 100410. [Google Scholar] [CrossRef]
- Shen, Y.; Li, W.; Zeng, Y.; Li, Z.; Chen, Y.; Zhang, J.; Zhao, H.; Feng, L.; Ma, D.; Mo, X.; et al. Chromosome-level and haplotype-resolved genome provides insight into the tetraploid hybrid origin of patchouli. Nat. Commun. 2022, 13, 3511. [Google Scholar] [CrossRef]
- Karunanithi, P.S.; Berrios, D.I.; Wang, S.; Davis, J.; Shen, T.; Fiehn, O.; Maloof, J.N.; Zerbe, P. The foxtail millet (Setaria italica) terpene synthase gene family. Plant J. 2020, 103, 781–800. [Google Scholar] [CrossRef]
- Falara, V.; Akhtar, T.A.; Nguyen, T.T.; Spyropoulou, E.A.; Bleeker, P.M.; Schauvinhold, I.; Matsuba, Y.; Bonini, M.E.; Schilmiller, A.L.; Last, R.L.; et al. The tomato terpene synthase gene family. Plant Physiol. 2011, 157, 770–789. [Google Scholar] [CrossRef]
- Martin, D.M.; Aubourg, S.; Schouwey, M.B.; Daviet, L.; Schalk, M.; Toub, O.; Lund, S.T.; Bohlmann, J. Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol. 2010, 10, 226. [Google Scholar] [CrossRef]
- Lian, Q.; Huettel, B.; Walkemeier, B.; Mayjonade, B.; Lopez-Roques, C.; Gil, L.; Roux, F.; Schneeberger, K.; Mercier, R. A pan-genome of 69 Arabidopsis thaliana accessions reveals a conserved genome structure throughout the global species range. Nat. Genet. 2024, 56, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-genome of wild and cultivated soybeans. Cell 2020, 182, 162–176.e13. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D.; et al. Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 2020, 182, 145–161.e23. [Google Scholar] [CrossRef]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558.e16. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, J.; Qi, Z.; Long, Y.; Pei, L.; Huang, X.; Grover, C.E.; Du, X.; Xia, C.; Wang, P.; et al. Genomic innovation and regulatory rewiring during evolution of the cotton genus Gossypium. Nat. Genet. 2022, 54, 1959–1971. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, P.T.; Kou, D.R.; Han, Y.C.; Fang, J.C.; Ni, J.P.; Jiang, B.; Wang, X.; Zhang, Y.J.; Wang, W.; et al. Terpene synthases in rice pan-genome and their responses to Chilo suppressalis larvae infesting. Front. Plant Sci. 2022, 13, 905982. [Google Scholar] [CrossRef]
- Sun, Y.; Xiao, W.; Wang, Q.N.; Wang, J.; Kong, X.D.; Ma, W.H.; Liu, S.X.; Ren, P.; Xu, L.N.; Zhang, Y.J. Multiple variation patterns of terpene synthases in 26 maize genomes. BMC Genom. 2023, 24, 46. [Google Scholar] [CrossRef]
- Hufford, M.B.; Seetharam, A.S.; Woodhouse, M.R.; Chougule, K.M.; Ou, S.; Liu, J.; Ricci, W.A.; Guo, T.; Olson, A.; Qiu, Y.; et al. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 2021, 373, 655–662. [Google Scholar] [CrossRef]
- Arce, C.M.; Besomi, G.; Glauser, G.; Turlings, T.C. Caterpillar-induced volatile emissions in cotton: The relative importance of damage and insect-derived factors. Front. Plant Sci. 2021, 12, 709858. [Google Scholar] [CrossRef]
- Clancy, M.V.; Mamin, M.; Flückiger, G.; Quijano-Medina, T.; Pérez-Niño, B.; Abdala-Roberts, L.; Turlings, T.C.; Bustos-Segura, C. Terpene chemotypes in Gossypium hirsutum (wild cotton) from the Yucatan Peninsula, Mexico. Phytochemistry 2023, 205, 113454. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, D.M.; Borges, M.; Laumann, R.A.; Caulfield, J.C.; Birkett, M.A.; Blassioli-Moraes, M.C. Inefficient weapon—The role of plant secondary metabolites in cotton defence against the boll weevil. Planta 2020, 252, 94. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Q.; Wu, X.M.; Ruan, J.X.; Hu, W.L.; Mao, Y.B.; Chen, X.Y.; Wang, L.J. Isolation and characterization of terpene synthases in cotton (Gossypium hirsutum). Phytochemistry 2013, 96, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.; Jin, Y.; Li, Y.; Nie, T.; Sun, L. Systematic identification of TPS genes in Gossypium and their characteristics in response to flooding stress. Front. Plant Sci. 2023, 14, 1126884. [Google Scholar] [CrossRef]
- Mehari, T.G.; Fang, H.; Feng, W.; Zhang, Y.; Umer, M.J.; Han, J.; Ditta, A.; Khan, M.K.; Liu, F.; Wang, K.; et al. Genome-wide identification and expression analysis of terpene synthases in Gossypium species in response to gossypol biosynthesis. Funct. Integr. Genom. 2023, 23, 197. [Google Scholar] [CrossRef]
- Huang, G.; Huang, J.Q.; Chen, X.Y.; Zhu, Y.X. Recent advances and future perspectives in cotton research. Annu. Rev. Plant Biol. 2021, 72, 437–462. [Google Scholar] [CrossRef]
- He, X.; Qi, Z.; Liu, Z.; Chang, X.; Zhang, X.; Li, J.; Wang, M. Pangenome analysis reveals transposon-driven genome evolution in cotton. BMC Biol. 2024, 22, 92. [Google Scholar] [CrossRef]
- Li, J.; Yuan, D.; Wang, P.; Wang, Q.; Sun, M.; Liu, Z.; Si, H.; Xu, Z.; Ma, Y.; Zhang, B.; et al. Cotton pan-genome retrieves the lost sequences and genes during domestication and selection. Genome Biol. 2021, 22, 119. [Google Scholar] [CrossRef]
- Huang, X.; Xiao, Y.; Köllner, T.G.; Zhang, W.; Wu, J.; Wu, J.; Guo, Y.; Zhang, Y. Identification and characterization of (E)-β-caryophyllene synthase and α/β-pinene synthase potentially involved in constitutive and herbivore-induced terpene formation in cotton. Plant Physiol. Bioch. 2013, 73, 302–308. [Google Scholar] [CrossRef]
- Huang, X.Z.; Chen, J.Y.; Xiao, H.J.; Xiao, Y.T.; Wu, J.; Wu, J.X.; Zhou, J.J.; Zhang, Y.J.; Guo, Y.Y. Dynamic transcriptome analysis and volatile profiling of Gossypium hirsutum in response to the cotton bollworm Helicoverpa armigera. Sci. Rep. 2015, 5, 11867. [Google Scholar] [CrossRef]
- Huang, X.; Kou, J.; Jing, W.; Han, X.; Liu, D.; Ghasemzadeh, S.; Sun, P.; Shi, W.; Zhang, Y. Transcriptomic and metabolomic reprogramming in cotton after Apolygus lucorum feeding implicated in enhancing recruitment of the parasitoid Peristenus spretus. J. Pest Sci. 2021, 95, 249–262. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, E.; Huang, X.; Kou, J.; Teng, D.; Lv, B.; Han, X.; Zhang, Y. Characterization of a novel insect-induced sesquiterpene synthase GbTPS1 based on the transcriptome of Gossypium barbadense feeding by cotton bollworm. Front. Plant Sci. 2022, 13, 898541. [Google Scholar] [CrossRef]
- Kou, J.; Teng, D.; Huang, X.; Lv, B.; Zhang, H.; Pan, H.; Zhang, Y. Overexpressing a cotton terpene synthase for (E)-β-ocimene biosynthesis in Nicotiana tabacum to recruit the parasitoid wasps. Ind. Crop. Prod. 2024, 222, 119476. [Google Scholar] [CrossRef]
- Wu, D.; Xie, L.; Sun, Y.; Huang, Y.; Jia, L.; Dong, C.; Shen, E.; Ye, C.Y.; Qian, Q.; Fan, L. A syntelog-based pan-genome provides insights into rice domestication and de-domestication. Genome Biol. 2023, 24, 179. [Google Scholar] [CrossRef]
- Wisecaver, J.H.; Borowsky, A.T.; Tzin, V.; Jander, G.; Kliebenstein, D.J.; Rokas, A. A global coexpression network approach for connecting genes to specialized metabolic pathways in plants. Plant Cell 2017, 29, 944–959. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, B.; Zheng, H.J.; Hu, Y.; Lu, G.; Yang, C.Q.; Chen, J.D.; Chen, J.J.; Chen, D.Y.; Zhang, L.; et al. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 2015, 5, 14139. [Google Scholar] [CrossRef]
- Wen, T.; Xu, X.; Ren, A.; Zhao, G.; Wu, J. Genome-wide identification of terpenoid synthase family genes in Gossypium hirsutum and functional dissection of its subfamily cadinene synthase A in gossypol synthesis. Front. Plant Sci. 2023, 14, 1162237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.P.; Zhang, J.L.; Sun, Z.R.; Liu, X.Y.; Shu, L.Z.; Wu, H.; Song, Y.; He, D.H. Genome-wide identification and characterization of terpene synthase genes in Gossypium hirsutum. Gene 2022, 828, 146462. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Z.; Wu, Y.; Zhang, Y.; Li, X.; Li, J.; Zhu, W.; Ma, Z.; Li, W. Terpene synthases GhTPS6 and GhTPS47 participate in resistance to Verticillium dahliae in upland cotton. Plant Physiol. Biochem. 2024, 213, 108798. [Google Scholar] [CrossRef]
- Tian, X.; Ruan, J.X.; Huang, J.Q.; Yang, C.Q.; Fang, X.; Chen, Z.W.; Hong, H.; Wang, L.J.; Mao, Y.B.; Lu, S.; et al. Characterization of gossypol biosynthetic pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E5410–E5418. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Jin, J.; Sarojam, R.; Ramachandran, S. A comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol. Evol. 2019, 11, 2078–2098. [Google Scholar] [CrossRef] [PubMed]
- Köllner, T.G.; Gershenzon, J.; Peters, R.J.; Zerbe, P.; Schmelz, E.A. The terpene synthase gene family in maize—A clarification of existing community nomenclature. BMC Genom. 2023, 24, 744. [Google Scholar] [CrossRef]
- Chen, X.Y.; Chen, Y.; Heinstein, P.; Davisson, V.J. Cloning, expression, and characterization of (+)-δ-cadinene synthase: A catalyst for cotton phytoalexin biosynthesis. Arch. Biochem. Biophys. 1995, 324, 255–266. [Google Scholar] [CrossRef]
- Chen, X.Y.; Wang, M.; Chen, Y.; Davisson, V.J.; Heinstein, P. Cloning and heterologous expression of a second (+)-δ-cadinene synthase from Gossypium arboreum. J. Nat. Prod. 1996, 59, 944–951. [Google Scholar] [CrossRef]
- Townsend, B.J.; Poole, A.; Blake, C.J.; Llewellyn, D.J. Antisense suppression of a (+)-δ-cadinene synthase gene in cotton prevents the induction of this defense response gene during bacterial blight infection but not its constitutive expression. Plant Physiol. 2005, 138, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Bao, T.; Kimani, S.; Li, Y.; Li, H.; Yang, S.; Zhang, J.; Wang, Q.; Wang, Z.; Ning, G.; Wang, L.; et al. Allelic variation of terpene synthases drives terpene diversity in the wild species of the Freesia genus. Plant Physiol. 2023, 192, 2419–2435. [Google Scholar] [CrossRef]
- He, J.; Fandino, R.A.; Halitschke, R.; Luck, K.; Köllner, T.G.; Murdock, M.H.; Ray, R.; Gase, K.; Knaden, M.; Baldwin, I.T.; et al. An unbiased approach elucidates variation in (S)-(+)-linalool, a context-specific mediator of a tri-trophic interaction in wild tobacco. Proc. Natl. Acad. Sci. USA 2019, 116, 14651–14660. [Google Scholar] [CrossRef] [PubMed]
- Köllner, T.G.; Held, M.; Lenk, C.; Hiltpold, I.; Turlings, T.C.; Gershenzon, J.; Degenhardt, J.R. A maize (E)-β-caryophyllene synthase implicated in indirect defense responses against herbivores is not expressed in most American maize varieties. Plant Cell 2008, 20, 482–494. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, L.; Liao, Y.; Li, J.; Tang, J.; Yang, Z. Formation of α-farnesene in tea (Camellia sinensis) leaves induced by herbivore-derived wounding and its effect on neighboring tea plants. Int. J. Mol. Sci. 2019, 20, 4151. [Google Scholar] [CrossRef]
- Röse, U.S.; Tumlinson, J.H. Volatiles released from cotton plants in response to Helicoverpa zea feeding damage on cotton flower buds. Planta 2004, 218, 824–832. [Google Scholar] [CrossRef]
- Liu, D.; Li, W.; An, X.; Ghasemzadeh, S.; Huang, X.; Chen, J.; Kou, J.; Sun, P.; Zhang, Y. Engineering Nicotiana tabacum for the de novo biosynthesis of DMNT to regulate orientation behavior of the parasitoid wasps Microplitis mediator. Pest Manag. Sci. 2021, 77, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Teng, D.; Liu, D.; Khashaveh, A.; Lv, B.; Sun, P.; Geng, T.; Cui, H.; Wang, Y.; Zhang, Y. Engineering DMNT emission in cotton enhances direct and indirect defense against mirid bugs. J. Adv. Res. 2024; in press. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. GGTREE: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Luo, X.; Kang, X.; Schönhuth, A. Phasebook: Haplotype-aware de novo assembly of diploid genomes from long reads. Genome Biol. 2021, 22, 299. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Rahmani, R.S.; Gugger, P.F.; Wang, M.; Li, H.; Zhang, Y.; Li, Z.; Wang, Q.; Van de Peer, Y.; Marchal, K.; et al. Distinct expression and methylation patterns for genes with different fates following a single whole-genome duplication in flowering plants. Mol. Biol. Evol. 2020, 37, 2394–2413. [Google Scholar] [CrossRef] [PubMed]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for visualization and analysis of biological networks. In Data Mining in Proteomics; Hamacher, M., Eisenacher, M., Stephan, C., Eds.; Humana Press: Totowa, NJ, USA, 2011; pp. 291–303. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Han, S.; Wang, M.; Ni, X.; Huang, X.; Zhang, Y. Pangenome Identification and Analysis of Terpene Synthase Gene Family Members in Gossypium. Int. J. Mol. Sci. 2024, 25, 9677. https://doi.org/10.3390/ijms25179677
Song Y, Han S, Wang M, Ni X, Huang X, Zhang Y. Pangenome Identification and Analysis of Terpene Synthase Gene Family Members in Gossypium. International Journal of Molecular Sciences. 2024; 25(17):9677. https://doi.org/10.3390/ijms25179677
Chicago/Turabian StyleSong, Yueqin, Shengjie Han, Mengting Wang, Xueqi Ni, Xinzheng Huang, and Yongjun Zhang. 2024. "Pangenome Identification and Analysis of Terpene Synthase Gene Family Members in Gossypium" International Journal of Molecular Sciences 25, no. 17: 9677. https://doi.org/10.3390/ijms25179677