Special Issue “The Role of Toll-Like Receptors (TLRs) in Infection and Inflammation 2.0”


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Author Contributions
Conflicts of Interest
References
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Taghavi, M.; Khosravi, A.; Mortaz, E.; Nikaein, D.; Athari, S.S. Role of pathogen-associated molecular patterns (PAMPS) in immune responses to fungal infections. Eur. J. Pharmacol. 2017, 808, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, D.G.; Green, T.L.; Lim, D.; Cho, K. Bacterial Pathogen–Associated Molecular Patterns Upregulate Human Glucocorticoid Receptor Expression in Peripheral Blood Mononuclear Cells. Shock 2022, 58, 393–399. [Google Scholar] [CrossRef]
- Eppensteiner, J.; Kwun, J.; Scheuermann, U.; Barbas, A.; Limkakeng, A.T.; Kuchibhatla, M.; Elster, E.A.; Kirk, A.D.; Lee, J. Damage- and pathogen-associated molecular patterns play differential roles in late mortality after critical illness. JCI Insight. 2019, 4, e127925. [Google Scholar] [CrossRef]
- Denk, S.; Perl, M.; Huber-Lang, M. Damage-and pathogen-associated molecular patterns and alarmins: Keys to sepsis? Eur. Surg. Res. 2012, 48, 171–179. [Google Scholar] [CrossRef]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518. [Google Scholar] [CrossRef]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- De Lorenzo, G.; Ferrari, S.; Cervone, F.; Okun, E. Extracellular DAMPs in Plants and Mammals: Immunity, Tissue Damage and Repair. Trends Immunol. 2018, 39, 937–950. [Google Scholar] [CrossRef]
- Mueller, C. Danger-associated molecular patterns and inflammatory bowel disease: Is there a connection? Dig. Dis. 2012, 30, 40–46. [Google Scholar] [CrossRef]
- Nofi, C.P.; Wang, P.; Aziz, M. Chromatin-Associated Molecular Patterns (CAMPs) in sepsis. Cell Death Dis. 2022, 13, 700. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2022, 180, 1044–1066. [Google Scholar] [CrossRef]
- Perales-Linares, R.; Navas-Martin, S. Toll-like receptor 3 in viral pathogenesis: Friend or foe? Immunology 2013, 140, 153–167. [Google Scholar] [CrossRef]
- Zheng, W.; Xu, Q.; Zhang, Y.; E, X.; Gao, W.; Zhang, M.; Zhai, W.; Rajkumar, R.S.; Liu, Z. Toll-like receptor-mediated innate immunity against herpesviridae infection: A current perspective on viral infection signaling pathways. Virol. J. 2020, 17, 192. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int. Immunopharmacol. 2020, 89, 107087. [Google Scholar] [CrossRef]
- Naqvi, I.; Giroux, N.; Olson, L.; Morrison, S.A.; Llanga, T.; Akinade, T.O.; Zhu, Y.; Zhong, Y.; Bose, S.; Arvai, S.; et al. DAMPs/PAMPs induce monocytic TLR activation and tolerance in COVID-19 patients; nucleic acid binding scavengers can counteract such TLR agonists. Biomaterials 2022, 283, 121393. [Google Scholar] [CrossRef]
- Gao, H.; Leaver, S.K.; Burke-Gaffney, A.; Finney, S.J. Severe sepsis and Toll-like receptors. Semin. Immunopathol. 2008, 30, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; E Huber, C. Bench-to-bedside review: Toll-like receptors and their role in septic shock. Crit. Care 2002, 6, 125–136. [Google Scholar] [CrossRef]
- Cook, D.N.; Pisetsky, D.S.; A Schwartz, D. Toll-like receptors in the pathogenesis of human disease. Nat. Immunol. 2004, 5, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Kiziltas, S. Toll-like receptors in pathophysiology of liver diseases. World J. Hepatol. 2016, 8, 1354–1369. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Planz, O. The Role of Toll-like Receptors (TLRs) and Their Related Signaling Pathways in Viral Infection and Inflammation. Int. J. Mol. Sci. 2023, 24, 6701. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Wu, K.-H.; Wu, H.-P. Unraveling the Complexities of Toll-like Receptors: From Molecular Mechanisms to Clinical Applications. Int. J. Mol. Sci. 2024, 25, 5037. [Google Scholar] [CrossRef]
- Billod, J.-M.; Lacetera, A.; Guzmán-Caldentey, J.; Martín-Santamaría, S. Computational Approaches to Toll-like Receptor 4 Modulation. Molecules 2016, 21, 994. [Google Scholar] [CrossRef]
- Murgueitio, M.S.; Rakers, C.; Frank, A.; Wolber, G. Balancing Inflammation: Computational Design of Small-Molecule Toll-like Receptor Modulators. Trends Pharmacol. Sci. 2016, 38, 155–168. [Google Scholar] [CrossRef]
- Klett, J.; Reeves, J.; Oberhauser, N.; Perez-Regidor, L.; Martin-Santamaria, S. Modulation of toll-like receptor 4. Insights from x-ray crystallography and molecular modeling. Curr. Top. Med. Chem. 2014, 14, 2672–2683. [Google Scholar] [CrossRef]
- Arciero, J.; Ermentrout, G.B.; Siggers, R.; Afrazi, A.; Hackam, D.; Vodovotz, Y.; Rubin, J. Modeling the interactions of bacteria and Toll-like receptor-mediated inflammation in necrotizing enterocolitis. J. Theor. Biol. 2013, 321, 83–99. [Google Scholar] [CrossRef]
- Farzan, M.; Mirzaei, Y.; Aiman, S.; Azadegan-Dehkordi, F.; Bagheri, N. Immunoinformatics-based multi-epitope vaccine design for the re-emerging monkeypox virus. Int. Immunopharmacol. 2023, 123, 110725. [Google Scholar] [CrossRef]
- Brooshghalan, S.E.; Sabahi, M.; Ebadi, S.A.; Sadeghian, Z.; Nayebi, A.M.; Haddadi, R. Silibinin chronic treatment in a rat model of Parkinson disease: A comprehensive in-vivo evaluation and in silico molecular modeling. Eur. J. Pharmacol. 2023, 941, 175517. [Google Scholar] [CrossRef] [PubMed]
- Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K. Toll-like Receptor Response to Human Immunodeficiency Virus Type 1 or Co-Infection with Hepatitis B or C Virus: An Overview. Int. J. Mol. Sci. 2023, 24, 9624. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Newcomb, C.W.; Carbonari, D.M.; Roy, J.A.; Torgersen, J.; Althoff, K.N.; Kitahata, M.M.; Reddy, K.R.; Lim, J.K.; Silverberg, M.J.; et al. Risk of HCC With Hepatitis B Viremia Among HIV/HBV-Coinfected Persons in North America. Hepatology 2021, 74, 1190–1202. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, Y.; Wu, J.; Chen, X.; Zou, W. HIV/HBV coinfection: Understanding the complex interactions and their impact on spontaneous HBV clearance, chronic liver damage, cirrhosis, and hepatocellular carcinoma. Aids Rev. 2024, 26, 032–040. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127 (Suppl. 1), S35–S50. [Google Scholar] [CrossRef]
- Puoti, M.; Prestini, K.; Putzolu, V.; Zanini, B.; Baiguera, C.; Antonini, M.G.; Pagani, P.; Airoldi, M.; Carosi, G. HIV/HCV co-infection: Natural history. Curr. Opin. HIV AIDS 2003, 17, 144–146. [Google Scholar]
- Maggiorella, M.T.; Sernicola, L.; Picconi, O.; Pizzi, E.; Belli, R.; Fulgenzi, D.; Rovetto, C.; Bruni, R.; Costantino, A.; Taffon, S.; et al. Epidemiological and molecular characterization of HBV and HCV infections in HIV-1-infected inmate population in Italy: A 2017–2019 multicenter cross-sectional study. Sci. Rep. 2023, 13, 14908. [Google Scholar] [CrossRef]
- Abravanel, F.; Raymond, S.; Pambrun, E.; Winnock, M.; Bonnard, P.; Sogni, P.; Trimoulet, P.; Dabis, F.; Salmon-Ceron, D.; Izopet, J.; et al. HIV-1 tropism and liver fibrosis in HIV–HCV co-infected patients. PLoS ONE 2012, 7, e50289. [Google Scholar] [CrossRef]
- Kim, A.Y.; Chung, R.T. Coinfection with HIV-1 and HCV—A one-two punch. Gastroenterology 2009, 137, 795–798. [Google Scholar] [CrossRef]
- Saracino, A.; Bruno, G.; Scudeller, L.; Punzi, G.; Lagioia, A.; Ladisa, N.; Monno, L.; Angarano, G. Does HIV-1 co-receptor tropism correlate with fibrosis progression in HIV/HCV co-infected patients? J. Clin. Virol. 2014, 59, 167–171. [Google Scholar] [CrossRef]
- Hu, J.; Liu, K.; Luo, J. HIV-HBV and HIV-HCV Coinfection and Liver Cancer Development. Cancer Treat. Res. 2019, 177, 231–250. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, G.; Berretta, M.; Pinzone, M.R.; Di Rosa, M.; Berretta, S.; Cunsolo, G.; Malaguarnera, M.; Cosentino, S.; De Paoli, P.; Schnell, J.M.; et al. Hepatocellular carcinoma in HIV positive patients. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1257–1270. [Google Scholar] [PubMed]
- El-Hage, N.; Podhaizer, E.M.; Sturgill, J.; Hauser, K.F. Toll-like receptor expression and activation in astroglia: Differential regulation by HIV-1 Tat, gp120, and morphine. Immunol. Investig. 2011, 40, 498–522. [Google Scholar] [CrossRef]
- Qin, X.; Yao, J.; Yang, F.; Nie, J.; Wang, Y.; Liu, P.C. Human immunodeficiency virus type 1 Nef in human monocyte-like cell line THP-1 expands treg cells via toll-like receptor 2. J. Cell. Biochem. 2011, 112, 3515–3524. [Google Scholar] [CrossRef]
- Sauter, D.; Kirchhoff, F. HIV replication: A game of hide and sense. Curr. Opin. HIV AIDS 2016, 11, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef]
- Hoshino, S.; Konishi, M.; Mori, M.; Shimura, M.; Nishitani, C.; Kuroki, Y.; Koyanagi, Y.; Kano, S.; Itabe, H.; Ishizaka, Y. HIV-1 Vpr induces TLR4/MyD88-mediated IL-6 production and reactivates viral production from latency. J. Leukoc. Biol. 2010, 87, 1133–1143. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, T.; Zhang, Y.; Luo, S.; Chen, H.; Chen, D.; Li, C.; Li, W. The reservoir of latent HIV. Front. Cell. Infect. Microbiol. 2022, 12, 945956. [Google Scholar] [CrossRef]
- Kreider, E.F.; Bar, K.J. HIV-1 Reservoir Persistence and Decay: Implications for Cure Strategies. Curr. HIV/AIDS Rep. 2022, 19, 194–206. [Google Scholar] [CrossRef]
- Li, S.; Moog, C.; Zhang, T.; Su, B. HIV reservoir: Antiviral immune responses and immune interventions for curing HIV infection. Chin. Med. J. 2022, 135, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Board, N.L.; Moskovljevic, M.; Wu, F.; Siliciano, R.F.; Siliciano, J.D. Engaging innate immunity in HIV-1 cure strategies. Nat. Rev. Immunol. 2022, 22, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Bricker, K.M.; Chahroudi, A.; Mavigner, M. New Latency Reversing Agents for HIV-1 Cure: Insights from Nonhuman Primate Models. Viruses 2021, 13, 1560. [Google Scholar] [CrossRef] [PubMed]
- Martinsen, J.T.; Gunst, J.D.; Højen, J.F.; Tolstrup, M.; Søgaard, O.S. The Use of Toll-like Receptor Agonists in HIV-1 Cure Strategies. Front. Immunol. 2020, 11, 1112. [Google Scholar] [CrossRef]
- Mantovani, S.; Oliviero, B.; Varchetta, S.; Renieri, A.; Mondelli, M.U. TLRs: Innate Immune Sentries against SARS-CoV-2 Infection. Int. J. Mol. Sci. 2023, 24, 8065. [Google Scholar] [CrossRef]
- Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K. An Overview of Recent Insights into the Response of TLR to SARS-CoV-2 Infection and the Potential of TLR Agonists as SARS-CoV-2 Vaccine Adjuvants. Viruses 2021, 13, 2302. [Google Scholar] [CrossRef]
- Isazadeh, A.; Heris, J.A.; Shahabi, P.; Mohammadinasab, R.; Shomali, N.; Nasiri, H.; Valedkarimi, Z.; Khosroshahi, A.J.; Hajazimian, S.; Akbari, M.; et al. Pattern-recognition receptors (PRRs) in SARS-CoV-2. Life Sci. 2023, 329, 121940. [Google Scholar] [CrossRef]
- Ghimire, R.; Shrestha, R.; Amaradhi, R.; Patton, T.; Whitley, C.; Chanda, D.; Liu, L.; Ganesh, T.; More, S.; Channappanavar, R. Toll-like receptor 7 (TLR7)-mediated antiviral response protects mice from lethal SARS-CoV-2 infection. bioRxiv 2023. [Google Scholar] [CrossRef]
- Cords, L.; Woost, R.; Kummer, S.; Brehm, T.T.; Kluge, S.; Schmiedel, S.; Jordan, S.; Lohse, A.W.; Altfeld, M.; Addo, M.M.; et al. Frequency of IRF5+ dendritic cells is associated with the TLR7-induced inflammatory cytokine response in SARS-CoV-2 infection. Cytokine 2023, 162, 156109. [Google Scholar] [CrossRef]
- Conti, P.; Younes, A. Coronavirus COV-19/SARS-CoV-2 affects women less than men: Clinical response to viral infection. J. Biol. Regul. Homeost. Agents 2020, 34, 339–343. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Rezaei, N. Role of Toll-like receptors in the pathogenesis of COVID-19. J. Med. Virol. 2021, 93, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H.; States, U. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Sariol, A.; Perlman, S. SARS-CoV-2 takes its Toll. Nat. Immunol. 2021, 22, 801–802. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sahanic, S.; Hilbe, R.; Dünser, C.; Tymoszuk, P.; Löffler-Ragg, J.; Rieder, D.; Trajanoski, Z.; Krogsdam, A.; Demetz, E.; Yurchenko, M.; et al. SARS-CoV-2 activates the TLR4/MyD88 pathway in human macrophages: A possible correlation with strong pro-inflammatory responses in severe COVID-19. Heliyon 2023, 9, e21893. [Google Scholar] [CrossRef] [PubMed]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 10, 8874339. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Herman, M.; Ciancanelli, M.J.; de Diego, R.P.; Sancho-Shimizu, V.; Abel, L.; Casanova, J.-L. TLR3 immunity to infection in mice and humans. Curr. Opin. Immunol. 2013, 25, 19–33. [Google Scholar] [CrossRef]
- Wang, P.; Liu, J.B.; Wang, X.; Meng, F.Z.; Xiao, Q.H.; Liu, L.; Zhu, J.; Hu, W.H.; Ho, W.Z. Activation of Toll-like receptor 3 inhibits HIV infection of human iPSC-derived microglia. J. Med. Virol. 2023, 95, e29217. [Google Scholar] [CrossRef]
- Choudhury, A.; Das, N.C.; Patra, R.; Mukherjee, S. In silico analyses on the comparative sensing of SARS-CoV-2 mRNA by the intracellular TLRs of humans. J. Med. Virol. 2021, 93, 2476–2486. [Google Scholar] [CrossRef]
- Croci, S.; Venneri, M.A.; Mantovani, S.; Fallerini, C.; Benetti, E.; Picchiotti, N.; Campolo, F.; Imperatore, F.; Palmieri, M.; Daga, S.; et al. The polymorphism L412F in TLR3 inhibits autophagy and is a marker of severe COVID-19 in males. Autophagy 2022, 18, 1662–1672. [Google Scholar] [CrossRef]
- Sung, P.-S.; Yang, S.-P.; Peng, Y.-C.; Sun, C.-P.; Tao, M.-H.; Hsieh, S.-L. CLEC5A and TLR2 are critical in SARS-CoV-2-induced NET formation and lung inflammation. J. Biomed. Sci. 2022, 29, 52. [Google Scholar] [CrossRef] [PubMed]
- Pedicillo, M.C.; De Stefano, I.S.; Zamparese, R.; Barile, R.; Meccariello, M.; Agostinone, A.; Villani, G.; Colangelo, T.; Serviddio, G.; Cassano, T.; et al. The Role of Toll-like Receptor-4 in Macrophage Imbalance in Lethal COVID-19 Lung Disease, and Its Correlation with Galectin-3. Int. J. Mol. Sci. 2023, 24, 13259. [Google Scholar] [CrossRef]
- Kircheis, R. In Silico Analyses Indicate a Lower Potency for Dimerization of TLR4/MD-2 as the Reason for the Lower Pathogenicity of Omicron Compared to Wild-Type Virus and Earlier SARS-CoV-2 Variants. Int. J. Mol. Sci. 2024, 25, 5451. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Planz, O. Could a Lower Toll-like Receptor (TLR) and NF-κB Activation Due to a Changed Charge Distribution in the Spike Protein Be the Reason for the Lower Pathogenicity of Omicron? Int. J. Mol. Sci. 2022, 23, 5966. [Google Scholar] [CrossRef]
- Colleselli, K.; Stierschneider, A.; Wiesner, C. An Update on Toll-like Receptor 2, Its Function and Dimerization in Pro- and Anti-Inflammatory Processes. Int. J. Mol. Sci. 2023, 24, 12464. [Google Scholar] [CrossRef]
- Gorecki, A.M.; Anyaegbu, C.C.; Anderton, R.S. TLR2 and TLR4 in Parkinson’s disease pathogenesis: The environment takes a toll on the gut. Transl. Neurodegener. 2021, 10, 47. [Google Scholar] [CrossRef]
- Dutta, D.; Jana, M.; Majumder, M.; Mondal, S.; Roy, A.; Pahan, K. Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nat. Commun. 2021, 12, 5382. [Google Scholar] [CrossRef]
- Sobrinho, H.M.d.R.; da Silva, D.J.; Gomides, L.F.; Dorta, M.L.; de Oliveira, M.A.P.; Ribeiro-Dias, F. TLR4 and TLR2 activation is differentially associated with age during Parkinson’s disease. Immunol. Investig. 2018, 47, 71–88. [Google Scholar] [CrossRef]
- Dutta, D.; Jana, M.; Paidi, R.K.; Majumder, M.; Raha, S.; Dasarathy, S.; Pahan, K. Tau fibrils induce glial inflammation and neuropathology via TLR2 in Alzheimer’s disease–related mouse models. J. Clin. Investig. 2023, 133, e161987. [Google Scholar] [CrossRef]
- Dallas, M.L.; Widera, D. TLR2 and TLR4-mediated inflammation in Alzheimer’s disease: Self-defense or sabotage? Neural Regen. Res. 2021, 16, 1552–1553. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lax, N.; Fainstein, N.; Nishri, Y.; Ben-Zvi, A.; Ben-Hur, T. Systemic microbial TLR2 agonists induce neurodegeneration in Alzheimer’s disease mice. J. Neuroinflamm. 2020, 17, 55. [Google Scholar] [CrossRef]
- Abarca-Merlin, D.M.; Martínez-Durán, J.A.; Medina-Pérez, J.D.; Rodríguez-Santos, G.; Alvarez-Arellano, L. From Immunity to Neurogenesis: Toll-like Receptors as Versatile Regulators in the Nervous System. Int. J. Mol. Sci. 2024, 25, 5711. [Google Scholar] [CrossRef] [PubMed]
- Squillace, S.; Salvemini, D. Toll-like receptor-mediated neuroinflammation: Relevance for cognitive dysfunctions. Trends Pharmacol. Sci. 2022, 43, 726–739. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef]
- Dubik, M.; Marczynska-Grzelak, J.; Sørensen, M.Z.; Dieu, R.S.; Rusin, D.; Schiöth, E.S.; Ramazani, B.; Belal, R.; Ojha, B.; Krieger, J.; et al. Synergistic Targeting of Innate Receptors TLR7 and NOD2 for Therapeutic Intervention in Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 7462. [Google Scholar] [CrossRef]
- Hernandez, J.; Schäffer, J.; Herden, C.; Pflieger, F.J.; Reiche, S.; Körber, S.; Kitagawa, H.; Welter, J.; Michels, S.; Culmsee, C.; et al. n-3 Polyunsaturated Fatty Acids Modulate LPS-Induced ARDS and the Lung–Brain Axis of Communication in Wild-Type versus Fat-1 Mice Genetically Modified for Leukotriene B4 Receptor 1 or Chemerin Receptor 23 Knockout. Int. J. Mol. Sci. 2023, 24, 13524. [Google Scholar] [CrossRef]
- Höpfinger, A.; Schmid, A.; Schweitzer, L.; Patz, M.; Weber, A.; Schäffler, A.; Karrasch, T. Regulation of Cathelicidin Antimicrobial Peptide (CAMP) Gene Expression by TNFα and cfDNA in Adipocytes. Int. J. Mol. Sci. 2023, 24, 15820. [Google Scholar] [CrossRef]
- Mylonas, K.S.; Peroulis, M.; Schizas, D.; Kapelouzou, A. MYD88 and Proinflammatory Chemokines in Aortic Atheromatosis: Exploring Novel Statin Effects. Int. J. Mol. Sci. 2023, 24, 9248. [Google Scholar] [CrossRef]
- Loaiza, R.; Fattahi, F.; Kalbitz, M.; Grailer, J.J.; Russell, M.W.; Jalife, J.; Valdivia, H.H.; Zetoune, F.S.; Ward, P.A. The Impact of Extracellular Histones and Absence of Toll-like Receptors on Cardiac Functional and Electrical Disturbances in Mouse Hearts. Int. J. Mol. Sci. 2024, 25, 8653. [Google Scholar] [CrossRef]
- Bosshart, H.; Heinzelmann, M. Targeting bacterial endotoxin: Two sides of a coin. Ann. N. Y. Acad. Sci. 2007, 1096, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Salaun, B.; Romero, P.; Lebecque, S. Toll-like receptors’ two-edged sword: When immunity meets apoptosis. Eur. J. Immunol. 2007, 37, 3311–3318. [Google Scholar] [CrossRef] [PubMed]
- Cario, E. Therapeutic impact of toll-like receptors on inflammatory bowel diseases: A multiple-edged sword. Inflamm. Bowel Dis. 2008, 14, 411–421. [Google Scholar] [CrossRef]
- Killeen, S.D.; Wang, J.H.; Andrews, E.J.; Redmond, H.P. Exploitation of the Toll-like receptor system in cancer: A doubled-edged sword? Br. J. Cancer 2006, 95, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.; Zipris, D. The Role of Toll-like receptor pathways in the mechanism of type 1 diabetes. Curr. Mol. Med. 2009, 9, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Manavalan, B.; Yoo, T.H.; Kim, S.G.; Choi, S. Roles of toll-like receptors in cancer: A double-edged sword for defense and offense. Arch. Pharmacal Res. 2012, 35, 1297–1316. [Google Scholar] [CrossRef]
- Marsh, B.J.; Stenzelpoore, M. Toll-like receptors: Novel pharmacological targets for the treatment of neurological diseases. Curr. Opin. Pharmacol. 2008, 8, 8–13. [Google Scholar] [CrossRef]
- Greene, C.; Connolly, R.; Brennan, D.; Laffan, A.; O’keeffe, E.; Zaporojan, L.; O’callaghan, J.; Thomson, B.; Connolly, E.; Argue, R.; et al. Blood–brain barrier disruption and sustained systemic inflammation in individuals with long COVID-associated cognitive impairment. Nat. Neurosci. 2024, 27, 421–432. [Google Scholar] [CrossRef]
- Su, W.; Ju, J.; Gu, M.; Wang, X.; Liu, S.; Yu, J.; Mu, D. SARS-CoV-2 envelope protein triggers depression-like behaviors and dysosmia via TLR2-mediated neuroinflammation in mice. J. Neuroinflamm. 2023, 20, 110. [Google Scholar] [CrossRef]
- Fontes-Dantas, F.L.; Fernandes, G.G.; Gutman, E.G.; De Lima, E.V.; Antonio, L.S.; Hammerle, M.B.; Mota-Araujo, H.P.; Colodeti, L.C.; Araújo, S.M.; Froz, G.M.; et al. SARS-CoV-2 Spike protein induces TLR4-mediated long-term cognitive dysfunction recapitulating post-COVID-19 syndrome in mice. Cell Rep. 2023, 42, 112189. [Google Scholar] [CrossRef]
- Kaushik, D.; Bhandari, R.; Kuhad, A. TLR4 as a therapeutic target for respiratory and neurological complications of SARS-CoV-2. Expert Opin. Ther. Targets 2021, 25, 491–508. [Google Scholar] [CrossRef]
- Frank, M.G.; Fleshner, M.; Maier, S.F. Exploring the immunogenic properties of SARS-CoV-2 structural proteins: PAMP:TLR signaling in the mediation of the neuroinflammatory and neurologic sequelae of COVID-19. Brain Behav. Immun. 2023, 111, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Burnett, F.N.; Coucha, M.; Bolduc, D.R.; Hermanns, V.C.; Heath, S.P.; Abdelghani, M.; Macias-Moriarity, L.Z.; Abdelsaid, M. SARS-CoV-2 Spike Protein Intensifies Cerebrovascular Complications in Diabetic hACE2 Mice through RAAS and TLR Signaling Activation. Int. J. Mol. Sci. 2023, 24, 16394. [Google Scholar] [CrossRef]
- Theoharides, T.C. Could SARS-CoV-2 Spike Protein Be Responsible for Long-COVID Syndrome? Mol. Neurobiol. 2022, 59, 1850–1861. [Google Scholar] [CrossRef] [PubMed]




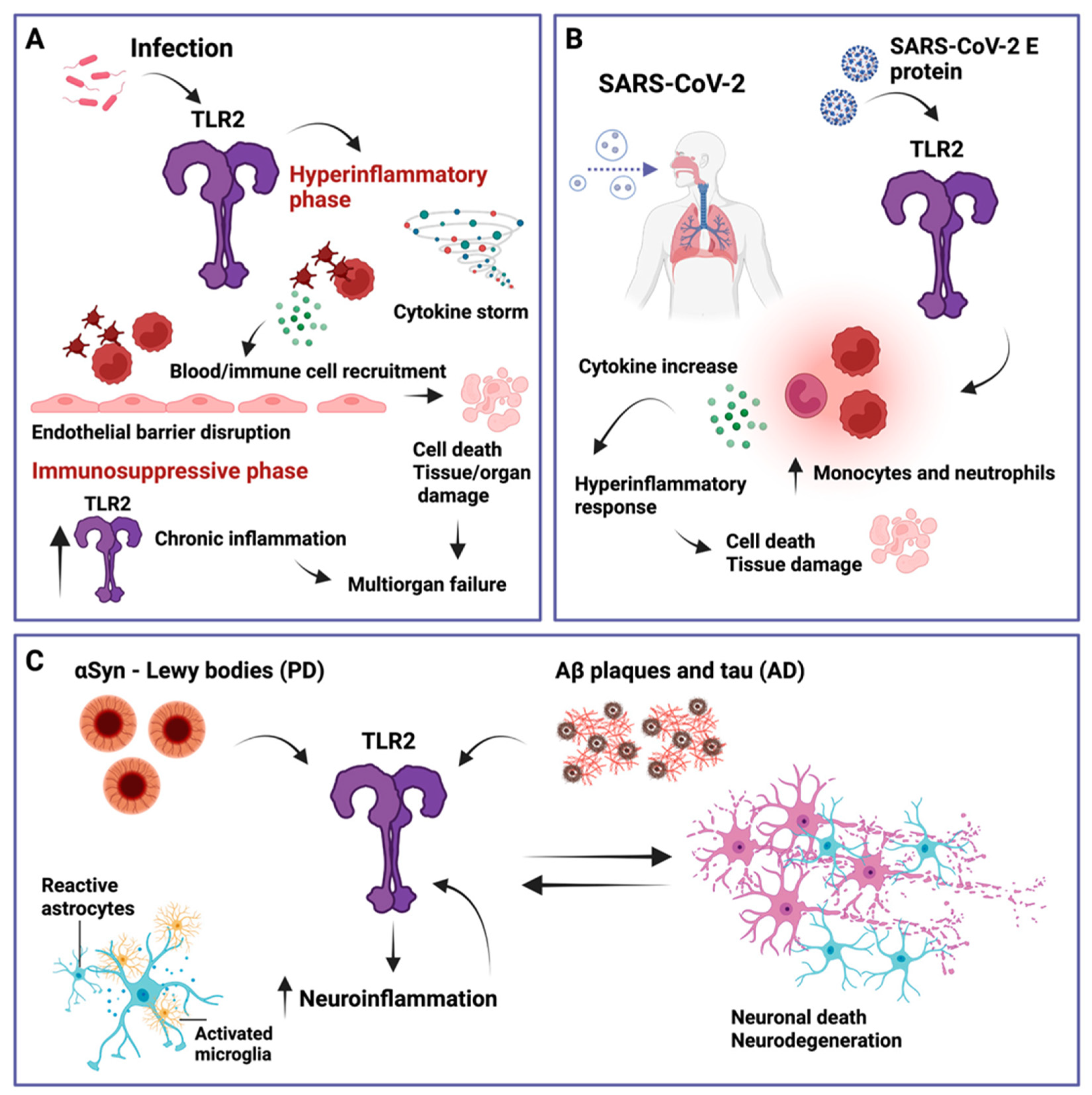
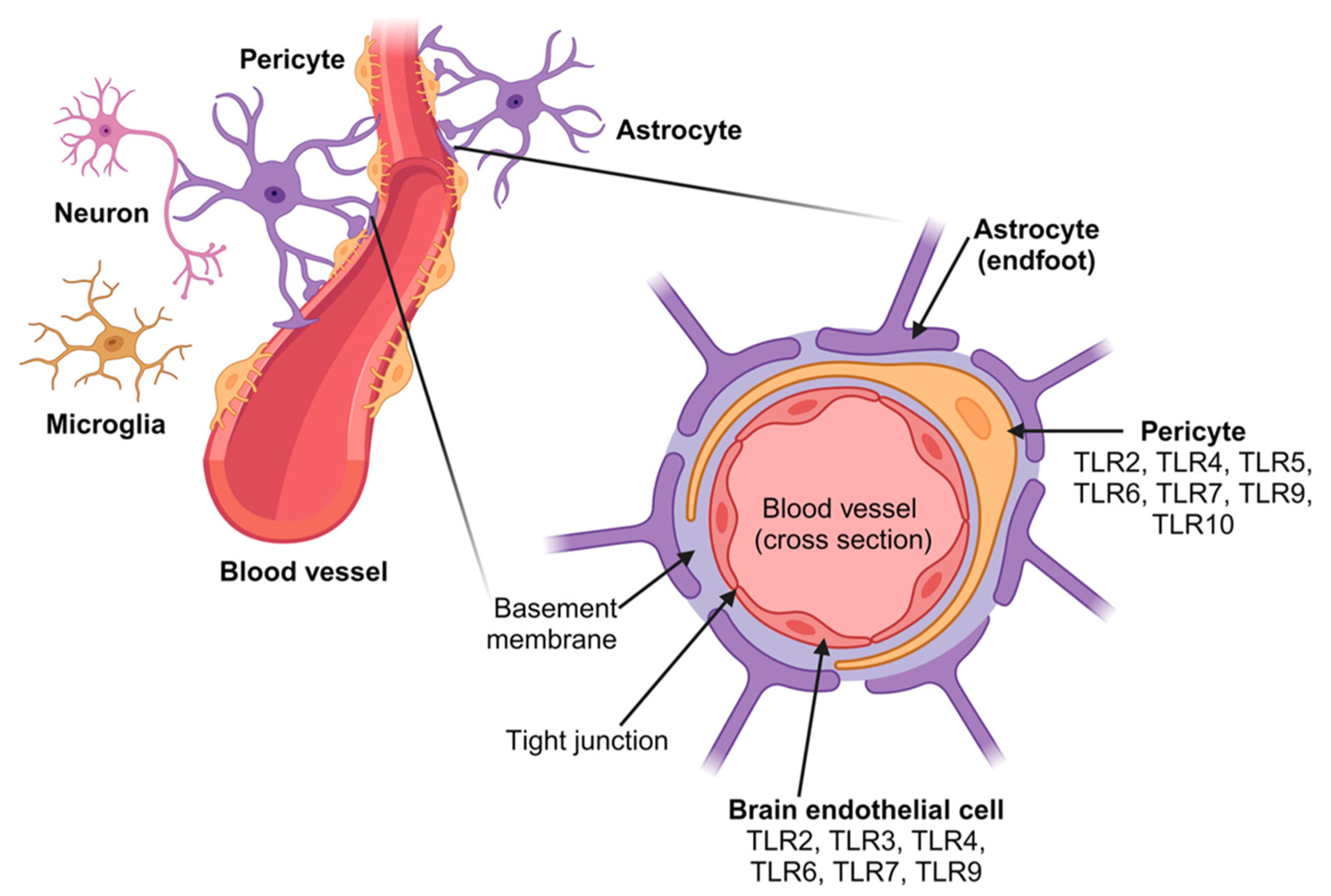
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kircheis, R.; Planz, O. Special Issue “The Role of Toll-Like Receptors (TLRs) in Infection and Inflammation 2.0”. Int. J. Mol. Sci. 2024, 25, 9709. https://doi.org/10.3390/ijms25179709
Kircheis R, Planz O. Special Issue “The Role of Toll-Like Receptors (TLRs) in Infection and Inflammation 2.0”. International Journal of Molecular Sciences. 2024; 25(17):9709. https://doi.org/10.3390/ijms25179709
Chicago/Turabian StyleKircheis, Ralf, and Oliver Planz. 2024. "Special Issue “The Role of Toll-Like Receptors (TLRs) in Infection and Inflammation 2.0”" International Journal of Molecular Sciences 25, no. 17: 9709. https://doi.org/10.3390/ijms25179709