
Insights into the Pathobiology of GM1 Gangliosidosis from Single-Nucleus Transcriptomic Analysis of CNS Cells in a Mouse Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Single-Nucleus Transcriptome Profiling Identified Cell Populations in the Brains of GM1 Gangliosidosis Mice
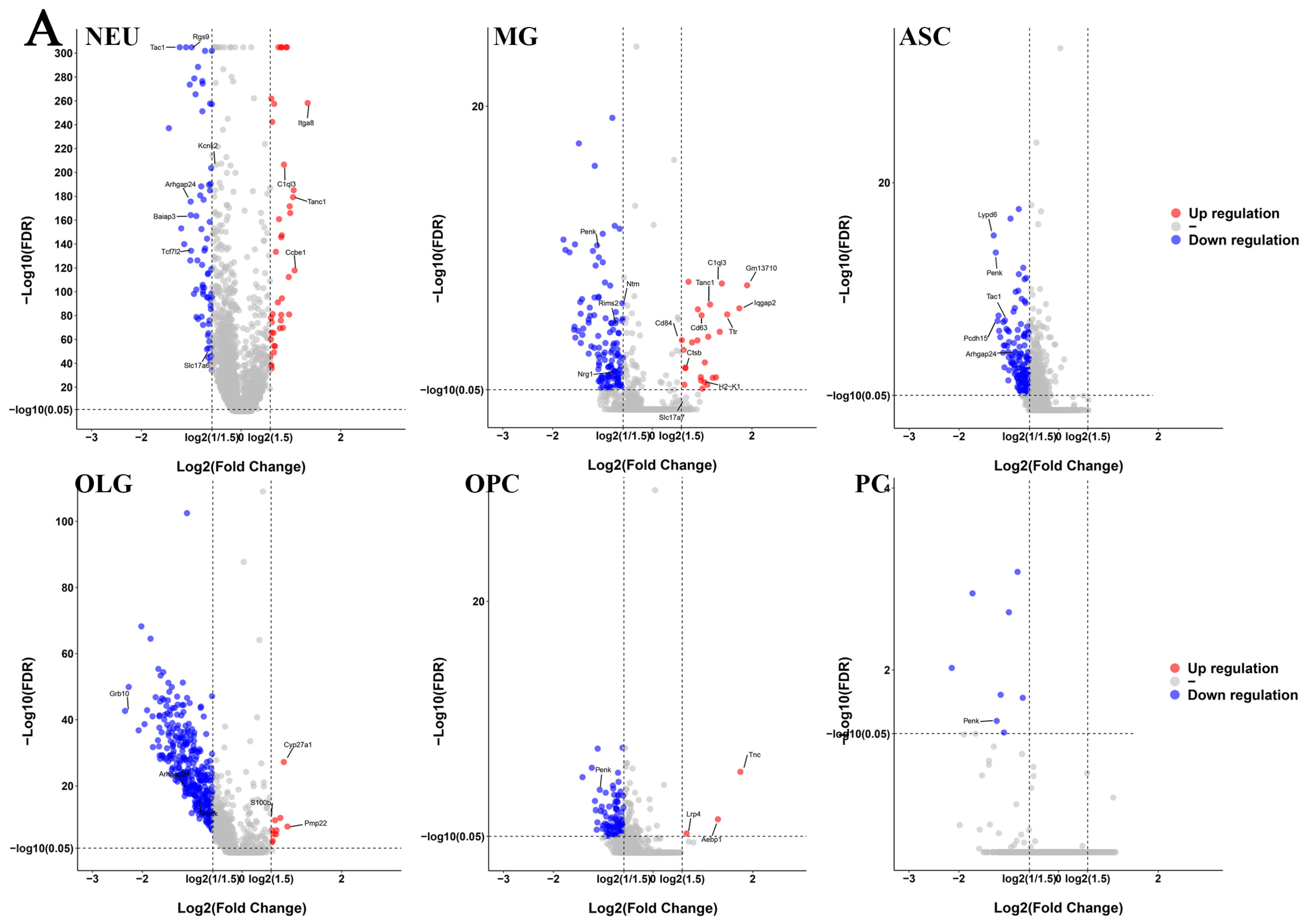
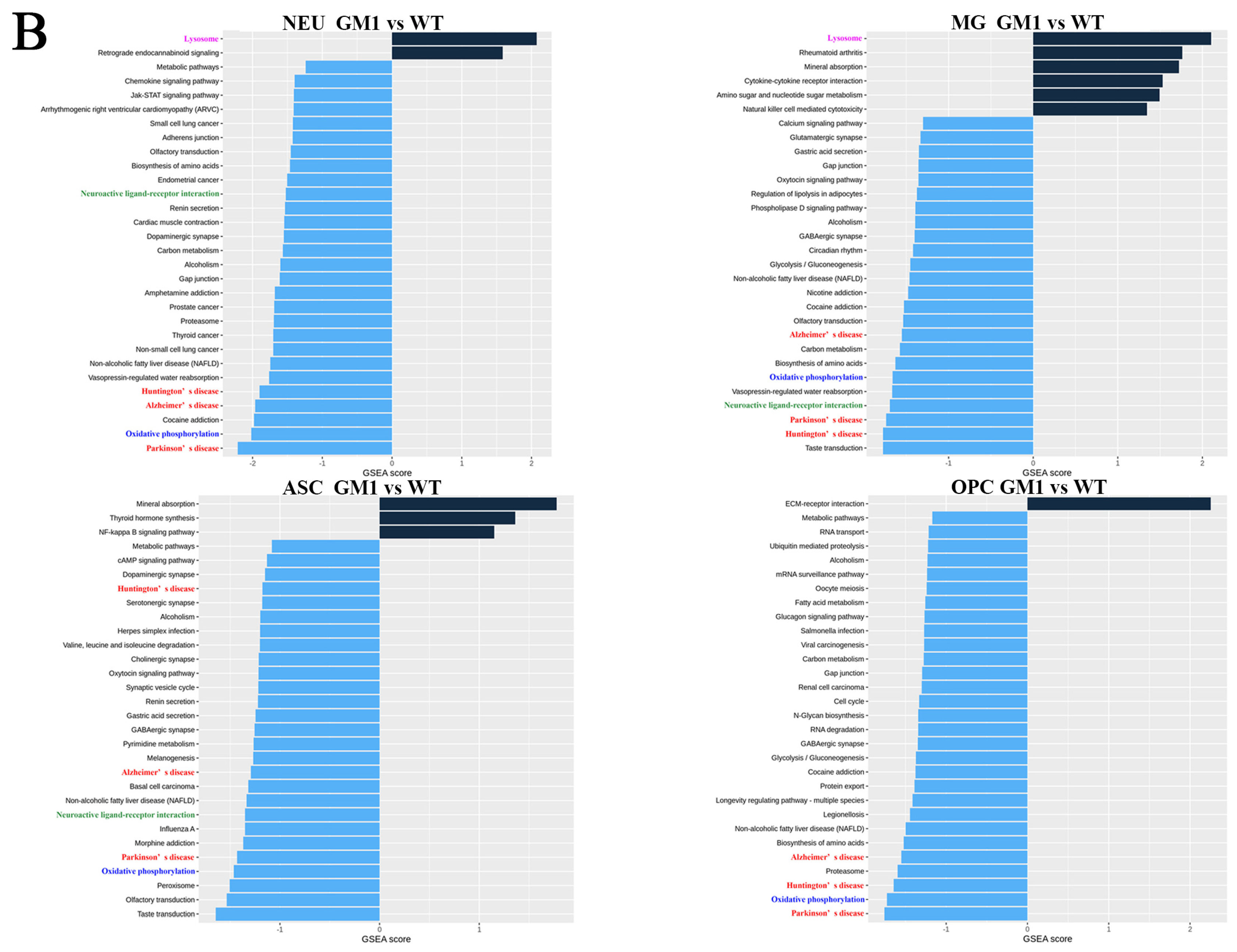
2.2. Differential Gene Expression and Functional Analyses Highlighted Abnormal Expression Patterns in Various Cell Types in GM1 Mice
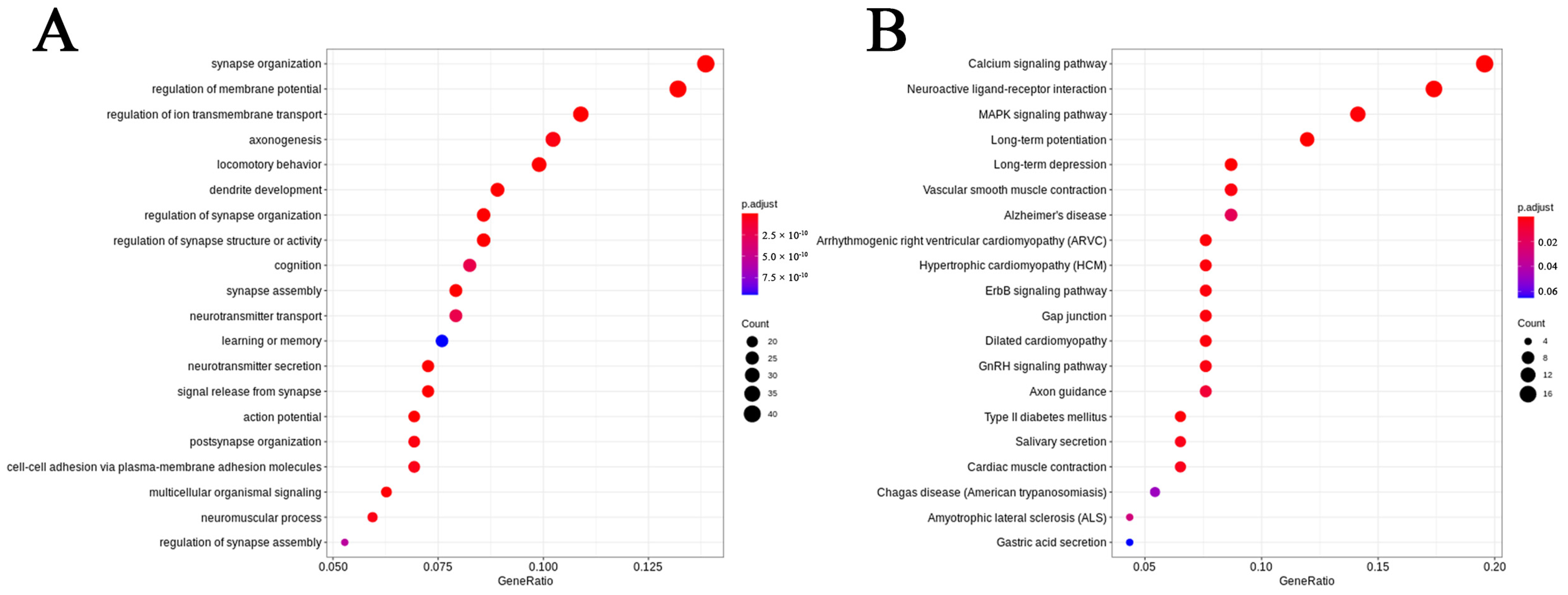
2.3. Altered Gene Expression in Neurons of GM1 Mice Induces Neurodegeneration-Related Changes
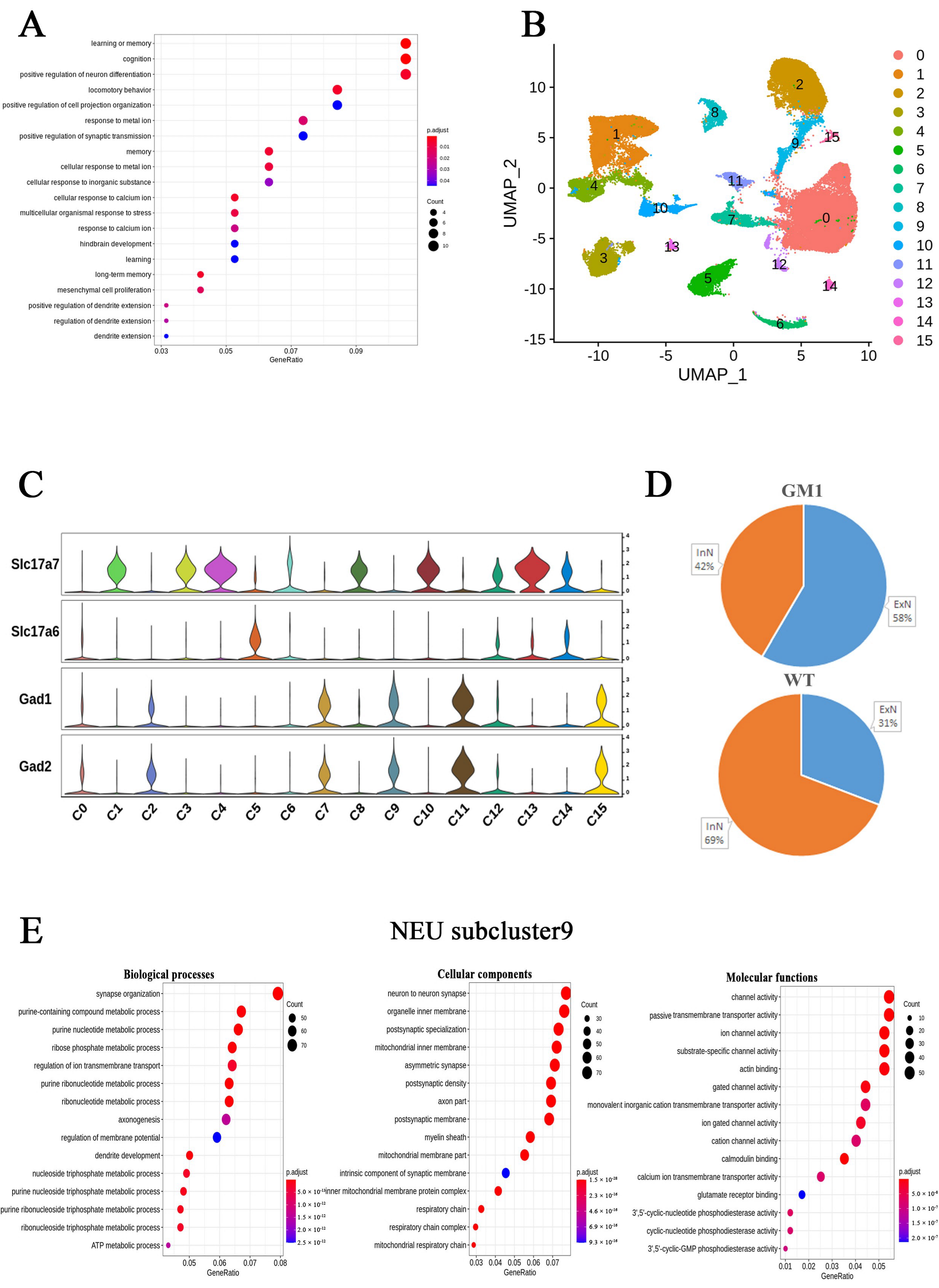
2.4. Microglia Exert Anti-Inflammatory Functions and Are Involved in the Regulation of Neuronal Death Signaling Pathways in GM1 Mice
2.5. Astrocytes Were Not Activated and Disrupted Synaptic Function Regulation in GM1 Mice
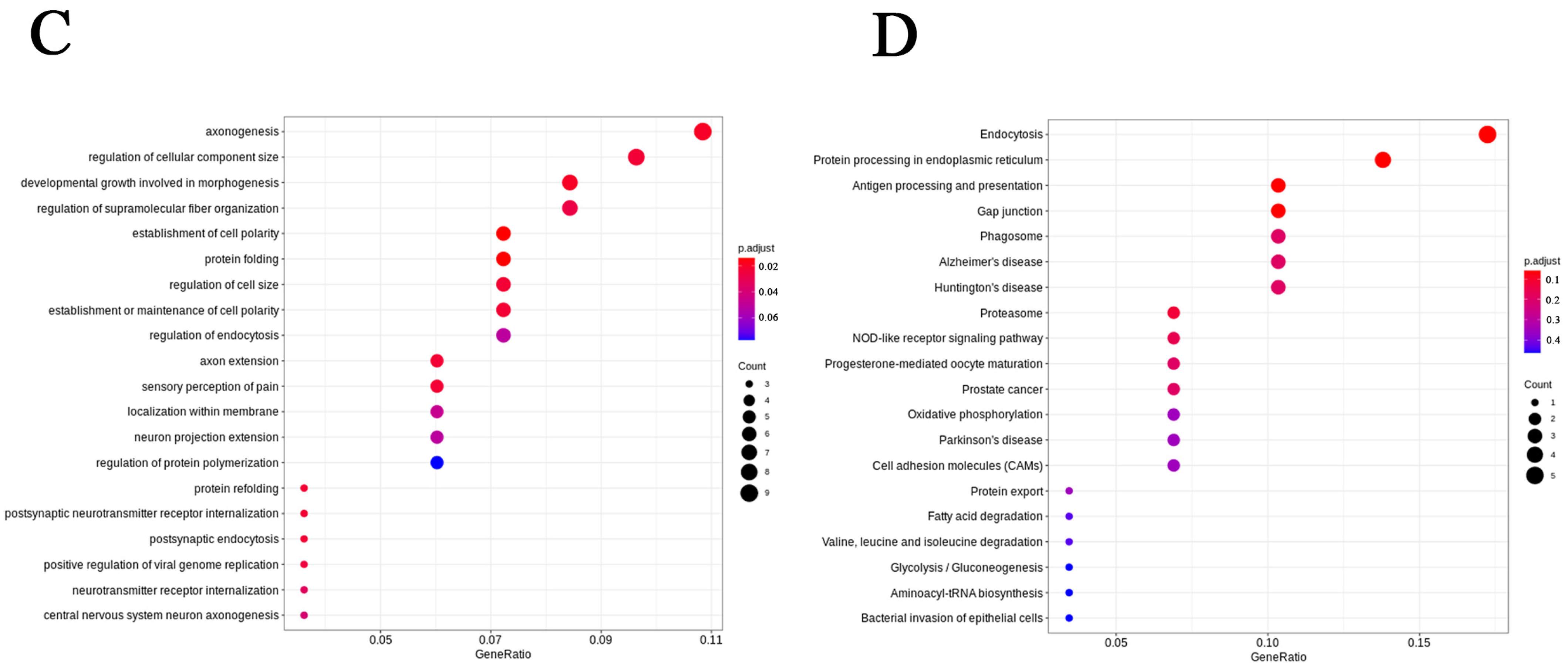
2.6. OLGs and OPCs Displayed Distinct Responses in GM1 Mice
2.7. Changes in Intercellular Communication in the CNS Revealed the Microenvironment in the GM1 Mouse Brain
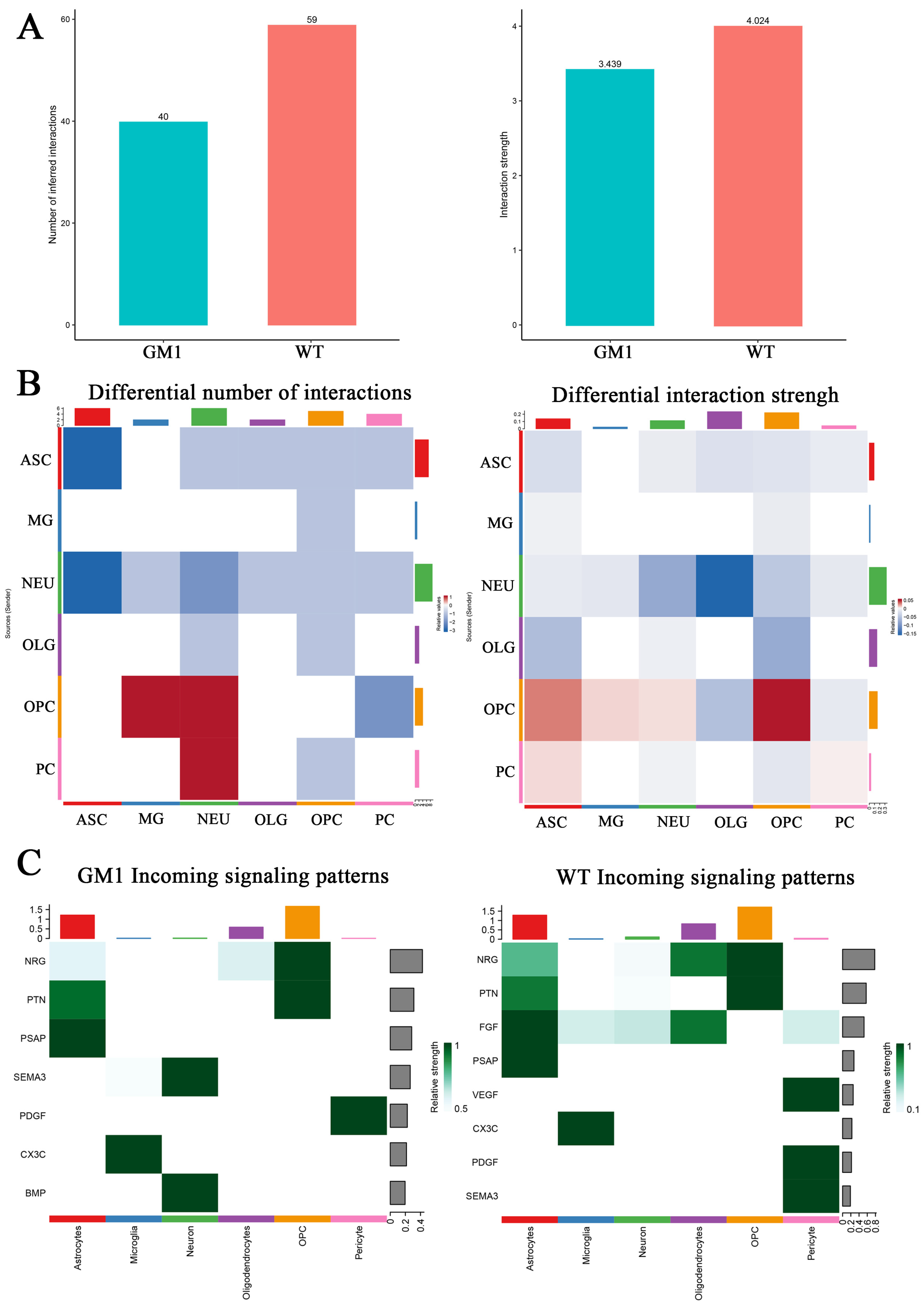
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Single-Nucleus RNA Sequencing
4.3. Data Processing and Quality Control
4.4. Clustering and Cell Type Identification
4.5. Differential Gene Expression and Pathway Analysis
4.6. Cell–Cell Communication Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Souza, P.; Farmer, C.; Johnston, J.; Han, S.T.; Adams, D.; Hartman, A.L.; Zein, W.; Huryn, L.A.; Solomon, B.; King, K.; et al. GM1 Gangliosidosis Type II: Results of a 10-Year Prospective Study. Genet. Genom. Med. 2024, 26, 101144. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z. Ganglioside GM1 and the Central Nervous System. Int. J. Mol. Sci. 2023, 24, 9558. [Google Scholar] [CrossRef]
- Liu, S.; Ma, W.; Feng, Y.; Zhang, Y.; Jia, X.; Tang, C.; Tang, F.; Wu, X.; Huang, Y. AAV9-coGLB1 Improves Lysosomal Storage and Rescues Central NervousSystem Inflammation in a Mutant Mouse Model of GM1 Gangliosidosis. Curr. Gene Ther. 2022, 22, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, E.-R.; Annunziata, I.; d’Azzo, A.; Platt, F.M.; Tifft, C.J.; Stepien, K.M. GM1 Gangliosidosis—A Mini-Review. Front. Genet. 2021, 12, 734878. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, K.; Fallahi, J.; Tabei, S.M.B.; Razban, V. Gene Therapy Approaches for GM1 Gangliosidosis: Focus on Animal and Cellular Studies. Cell Biochem. Amp Funct. 2023, 41, 1093–1105. [Google Scholar] [CrossRef]
- Foster, D.; Williams, L.; Arnold, N.; Larsen, J. Therapeutic Developments for Neurodegenerative GM1 Gangliosidosis. Front. Neurosci. 2024, 18, 1392683. [Google Scholar] [CrossRef]
- Ghosh, A.; Héron, B.; Donald, A.; Chang, H.; Furness, L.; Movsesyan, N.; Ravelli, C.; Jones, S.; Wang, R.Y. Safety and Preliminary Efficacy of LYS-GM101 Gene Therapy in Patients with GM1 Gangliosidosis: Results of a Phase I/II Open-Label Clinical Trial. Mol. Genet. Metab. 2024, 141, 107839. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal Storage Diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Lysosomal Glycosphingolipid Storage Diseases. Annu. Rev. Biochem. 2019, 28, 461–485. [Google Scholar] [CrossRef]
- Cougnoux, A.; Yerger, J.C.; Fellmeth, M.; Serra-Vinardell, J.; Martin, K.; Navid, F.; Iben, J.; Wassif, C.A.; Cawley, N.X.; Porter, F.D. Single Cell Transcriptome Analysis of Niemann–Pick Disease, Type C1 Cerebella. Int. J. Mol. Sci. 2020, 21, 5368. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W.; et al. Single-Cell Profiling of the Developing Mouse Brain and Spinal Cord with Split-Pool Barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Zhang, Y.; Li, Y.; Lin, X.; Guo, Z.; Hu, H.; Shao, W.; Yu, G.; Zheng, F.; Cai, P.; et al. Single-Cell RNA-Sequencing of Cellular Heterogeneity and Pathogenic Mechanisms in Paraquat-Induced Parkinson’s Disease with Depression. Ecotoxicol. Environ. Saf. 2024, 273, 116169. [Google Scholar] [CrossRef] [PubMed]
- Lv, N.; Wang, Y.; Liu, Y.; Tang, J.; Lei, Q.; Wang, Y.; Wei, H. Decreased Microglia in Pax2 Mutant Mice Leads to Impaired Learning and Memory. ACS Chem. Neurosci. 2022, 13, 2490–2502. [Google Scholar] [CrossRef]
- Niemela, V.; Landtblom, A.; Nyholm, D.; Kneider, M.; Constantinescu, R.; Paucar, M.; Svenningsson, P.; Abujrais, S.; Burman, J.; Shevchenko, G.; et al. Proenkephalin Decreases in Cerebrospinal Fluid with Symptom Progression of Huntington’s Disease. Mov. Disord. 2021, 36, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, S.; Vaillancourt, M.; Hébert, S.S.; Drolet, G.; Samadi, P. Striatal Pre-Enkephalin Overexpression Improves Huntington’s Disease Symptoms in the R6/2 Mouse Model of Huntington’s Disease. PLoS ONE 2013, 8, e75099. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Ohta, Y. RSK/GSK3–Mediated Phosphorylation of FilGAP Regulates Chemotactic Cancer Invasion. PNAS Nexus 2024, 3, pgae071. [Google Scholar] [CrossRef]
- Yang, C.; Pang, Y.; Huang, Y.; Ye, F.; Chen, X.; Gao, Y.; Zhang, C.; Yao, L.; Gao, J. Single-Cell Transcriptomics Identifies Premature Aging Features of TERC-Deficient Mouse Brain and Bone Marrow. Geroscience 2022, 44, 2139–2155. [Google Scholar] [CrossRef]
- Little, K.; Singh, A.; Del Marco, A.; Llorián-Salvador, M.; Vargas-Soria, M.; Turch-Anguera, M.; Solé, M.; Bakker, N.; Scullion, S.; Comella, J.X.; et al. Disruption of Cortical Cell Type Composition and Function Underlies Diabetes-Associated Cognitive Decline. Diabetologia 2023, 66, 1557–1575. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.-E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia States and Nomenclature: A Field at Its Crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Maksour, S.; Ooi, L. Innovations Advancing Our Understanding of Microglia in Alzheimer’s Disease: From in Vitro to in Vivo Models. J. Neurochem. 2023, jnc.15885. [Google Scholar] [CrossRef] [PubMed]
- Edland, S.; Hernandez, F.; Nasamran, C.; Fisch, K.; White, L. GWAS OF BRAIN ENDOPHENOTYPES: ASSOCIATION OF AUTOPSY-BASED LEWY BODY COUNTS WITH MOVEMENT DISORDER RISK LOCI. Innov. Aging 2023, 7, 688. [Google Scholar] [CrossRef]
- Liakath-Ali, K.; Refaee, R.; Südhof, T.C. Cartography of Teneurin and Latrophilin Expression Reveals Spatiotemporal Axis Heterogeneity in the Mouse Hippocampus during Development. PLoS Biol. 2024, 22, e3002599. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Siddiqui, T.J.; Padmanabhan, N.; Wallin, J.; Zhang, C.; Karimi, B.; Rieke, F.; Craig, A.M.; Wong, R.O.; Hoon, M. LRRTM4: A Novel Regulator of Presynaptic Inhibition and Ribbon Synapse Arrangements of Retinal Bipolar Cells. Neuron 2020, 105, 1007–1017.e5. [Google Scholar] [CrossRef]
- Humphries, C.E.; Kohli, M.A.; Nathanson, L.; Whitehead, P.; Beecham, G.; Martin, E.; Mash, D.C.; Pericak-Vance, M.A.; Gilbert, J. Integrated Whole Transcriptome and DNA Methylation Analysis Identifies Gene Networks Specific to Late-Onset Alzheimer’s Disease. JAD 2015, 44, 977–987. [Google Scholar] [CrossRef]
- Ding, Z.; Dai, C.; Zhong, L.; Liu, R.; Gao, W.; Zhang, H.; Yin, Z. Neuregulin-1 Converts Reactive Astrocytes toward Oligodendrocyte Lineage Cells via Upregulating the PI3K-AKT-mTOR Pathway to Repair Spinal Cord Injury. Biomed. Pharmacother. 2021, 134, 111168. [Google Scholar] [CrossRef] [PubMed]
- Bauch, J.; Faissner, A. The Extracellular Matrix Proteins Tenascin-C and Tenascin-R Retard Oligodendrocyte Precursor Maturation and Myelin Regeneration in a Cuprizone-Induced Long-Term Demyelination Animal Model. Cells 2022, 11, 1773. [Google Scholar] [CrossRef]
- Zhou, T.; Ma, Y.; Tang, J.; Guo, F.; Dong, M.; Wei, Q. Modulation of IGF1R Signaling Pathway by GIGYF1 in High Glucose-Induced SHSY-5Y Cells. DNA Cell Biol. 2018, 37, 1044–1054. [Google Scholar] [CrossRef]
- Ma, L.; Wei, Q.; Deng, H.; Zhang, Q.; Li, G.; Tang, N.; Xie, J.; Chen, Y. Growth Factor Receptor-Bound Protein 10-Mediated Negative Regulation of the Insulin-Like Growth Factor-1 Receptor-Activated Signalling Pathway Results in Cognitive Disorder in Diabetic Rats. J. Neuroendocrinol. 2013, 25, 626–634. [Google Scholar] [CrossRef]
- Rha, A.K.; Maguire, A.S.; Martin, D.R. GM1 Gangliosidosis: Mechanisms and Management. Appl. Clin. Genet. 2021, 14, 209–233. [Google Scholar] [CrossRef]
- Allen, N.J.; Lyons, D.A. Glia as Architects of Central Nervous System Formation and Function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Czopka, T. Myelination-Independent Functions of Oligodendrocyte Precursor Cells in Health and Disease. Nat. Neurosci. 2023, 26, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.A.; McBride, M.R.; Baker, H.J.; Christian, S.T. Fluorescence Polarization Analysis, Lipid Composition, and Na+, K+ -ATPase Kinetics of Synaptosomal Membranes in Feline GM1 and GM2 Gangliosidosis. J. Neurochem. 1985, 44, 947–956. [Google Scholar] [CrossRef]
- Soukup, S.-F.; Vanhauwaert, R.; Verstreken, P. Parkinson’s Disease: Convergence on Synaptic Homeostasis. EMBO J. 2018, 37, e98960. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, Calcium and Mitochondria: A Triad in Synaptic Neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Kajihara, R.; Numakawa, T.; Odaka, H.; Yaginuma, Y.; Fusaki, N.; Okumiya, T.; Furuya, H.; Inui, S.; Era, T. Novel Drug Candidates Improve Ganglioside Accumulation and Neural Dysfunction in GM1 Gangliosidosis Models with Autophagy Activation. Stem Cell Rep. 2020, 14, 909–923. [Google Scholar] [CrossRef]
- Singer, H.S.; Coyle, J.T.; Weaver, D.L.; Kawamura, N.; Baker, H.J. Neurotransmitter Chemistry in Feline GM1 Gangliosidosis: A Model for Human Ganglioside Storage Disease. Ann. Neurol. 1982, 12, 37–41. [Google Scholar] [CrossRef]
- Frase, S.; Löffler, F.; Hosp, J.A. Enhancing Post-Stroke Rehabilitation and Preventing Exo-Focal Dopaminergic Degeneration in Rats—A Role for Substance P. Int. J. Mol. Sci. 2022, 23, 3848. [Google Scholar] [CrossRef]
- Bissonnette, S.; Muratot, S.; Vernoux, N.; Bezeau, F.; Calon, F.; Hébert, S.S.; Samadi, P. The Effect of Striatal Pre-enkephalin Overexpression in the Basal Ganglia of the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Mouse Model of Parkinson’s Disease. Eur. J. Neurosci. 2014, 40, 2406–2416. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.; Lee, H.; Shin, E.; Kang, H.; Jeon, J.; Youn, B. Baiap3 Regulates Depressive Behaviors in Mice via Attenuating Dense Core Vesicle Trafficking in Subsets of Prefrontal Cortex Neurons. Neurobiol. Stress 2022, 16, 100423. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Talantova, M.; McKercher, S.R.; Lipton, S.A. Novel Therapeutic Approach for Excitatory/Inhibitory Imbalance in Neurodevelopmental and Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 701–721. [Google Scholar] [CrossRef]
- Von Scheibler, E.N.M.M.; Van Eeghen, A.M.; De Koning, T.J.; Kuijf, M.L.; Zinkstok, J.R.; Müller, A.R.; Van Amelsvoort, T.A.M.J.; Boot, E. Parkinsonism in Genetic Neurodevelopmental Disorders: A Systematic Review. Mov. Disord. Clin. Pract. 2023, 10, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M. Central Nervous System Inflammation Is a Hallmark of Pathogenesis in Mouse Models of GM1 and GM2 Gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef]
- Liu, S.; Feng, Y.; Huang, Y.; Jiang, X.; Tang, C.; Tang, F.; Zeng, C.; Liu, L. A GM1 Gangliosidosis Mutant Mouse Model Exhibits Activated Microglia And. Exp. Biol. Med. 2021, 246, 1330–1341. [Google Scholar] [CrossRef]
- Feltes, M.; Gale, S.E.; Moores, S.; Ory, D.S.; Schaffer, J.E. Monitoring the Itinerary of Lysosomal Cholesterol in Niemann-Pick Type C1-Deficient Cells after Cyclodextrin Treatment. J. Lipid Res. 2020, 61, 403–412. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Z.; Zhang, K.; Zhu, J.; Jin, T. FGF/FGFR System in the Central Nervous System Demyelinating Disease: Recent Progress and Implications for Multiple Sclerosis. CNS Neurosci. Ther. 2023, 29, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Q.; Cai, H.; He, Z.; Wang, H.; Chen, J.; Zheng, Z.; Yin, J.; Liao, Z.; Xu, H.; et al. FGF1 Improves Functional Recovery through Inducing PRDX1 to Regulate Autophagy and Anti-ROS after Spinal Cord Injury. J. Cell. Mol. Med. 2018, 22, 2727–2738. [Google Scholar] [CrossRef]
- Huang, W.-C.; Kuo, H.-S.; Tsai, M.-J.; Ma, H.; Chiu, C.-W.; Huang, M.-C.; Yang, L.-H.; Chang, P.-T.; Lin, Y.-L.; Kuo, W.-C.; et al. Adenoassociated Virusmediated Human Acidic Fibroblast Growth Factor Expression Promotes Functional Recovery of Spinal Cordcontused Rats. J. Gene Med. 2011, 13, 283–289. [Google Scholar] [CrossRef]
- Furusho, M.; Dupree, J.L.; Nave, K.-A.; Bansal, R. Fibroblast Growth Factor Receptor Signaling in Oligodendrocytes Regulates Myelin Sheath Thickness. J. Neurosci. 2012, 32, 6631–6641. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial Reconstruction of Single-Cell Gene Expression Data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast Unfolding of Communities in Large Networks. J. Stat. Mech. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-Based Analysis of Lung Single-Cell Sequencing Reveals a Transitional Profibrotic Macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Draghici, S.; Khatri, P.; Tarca, A.L.; Amin, K.; Done, A.; Voichita, C.; Georgescu, C.; Romero, R. A Systems Biology Approach for Pathway Level Analysis. Genome Res. 2007, 17, 1537–1545. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Xie, T.; Huang, Y. Insights into the Pathobiology of GM1 Gangliosidosis from Single-Nucleus Transcriptomic Analysis of CNS Cells in a Mouse Model. Int. J. Mol. Sci. 2024, 25, 9712. https://doi.org/10.3390/ijms25179712
Liu S, Xie T, Huang Y. Insights into the Pathobiology of GM1 Gangliosidosis from Single-Nucleus Transcriptomic Analysis of CNS Cells in a Mouse Model. International Journal of Molecular Sciences. 2024; 25(17):9712. https://doi.org/10.3390/ijms25179712
Chicago/Turabian StyleLiu, Sichi, Ting Xie, and Yonglan Huang. 2024. "Insights into the Pathobiology of GM1 Gangliosidosis from Single-Nucleus Transcriptomic Analysis of CNS Cells in a Mouse Model" International Journal of Molecular Sciences 25, no. 17: 9712. https://doi.org/10.3390/ijms25179712
APA StyleLiu, S., Xie, T., & Huang, Y. (2024). Insights into the Pathobiology of GM1 Gangliosidosis from Single-Nucleus Transcriptomic Analysis of CNS Cells in a Mouse Model. International Journal of Molecular Sciences, 25(17), 9712. https://doi.org/10.3390/ijms25179712