
Glucose-Dependent Insulinotropic Polypeptide Inhibits AGE-Induced NADPH Oxidase-Derived Oxidative Stress Generation and Foam Cell Formation in Macrophages Partly via AMPK Activation †
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
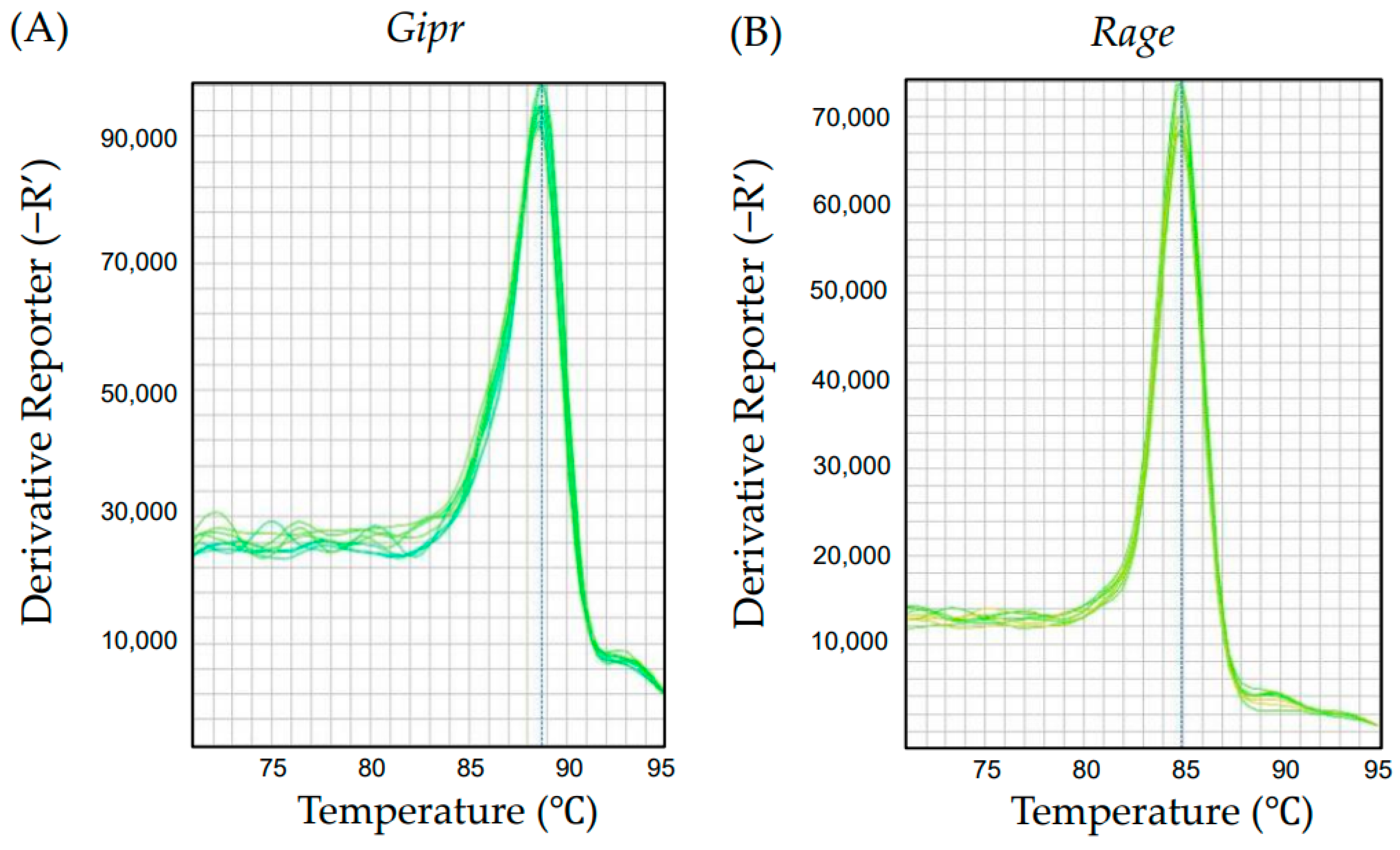
2.1. Gene Expression of Gipr and Rage in Human Monocyte-Derived U937 Cells
2.2. [D-Ala2]GIP(1–42) Inhibited the AGE-Induced Foam Cell Formation of and Intracellular Reactive Oxygen Species (ROS) Generation in Human U937 Cells
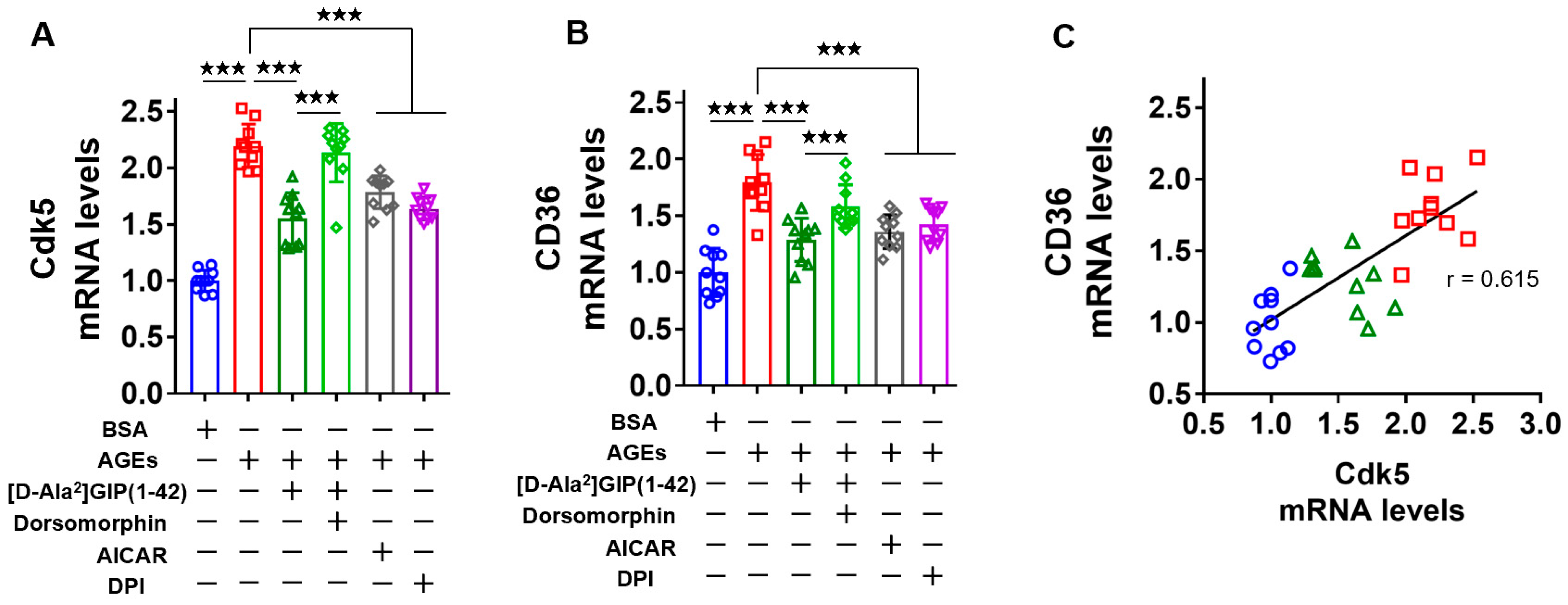
2.3. [D-Ala2]GIP(1–42) Inhibited Cdk5 and CD36 Gene Expression Levels in AGE-Exposed U937 Cells
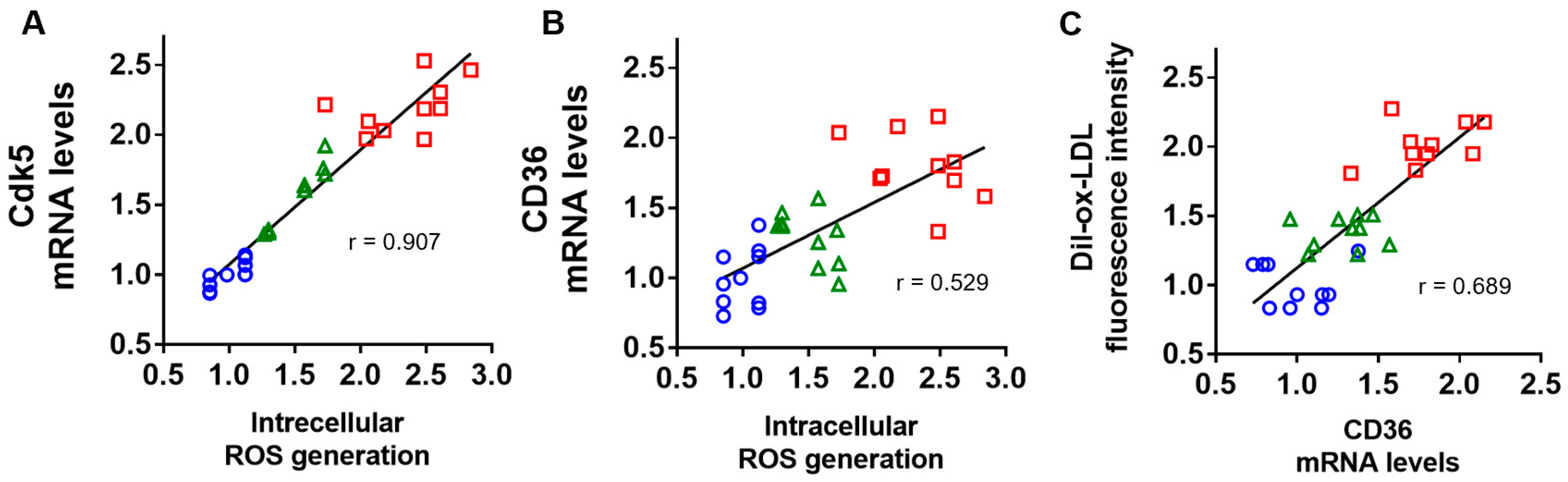
2.4. Correlation of Intracellular ROS Generation with Cdk5 and CD36 Gene Expression, and Association of CD36 mRNA Levels with Dil-ox-LDL Uptake into U937 Cells
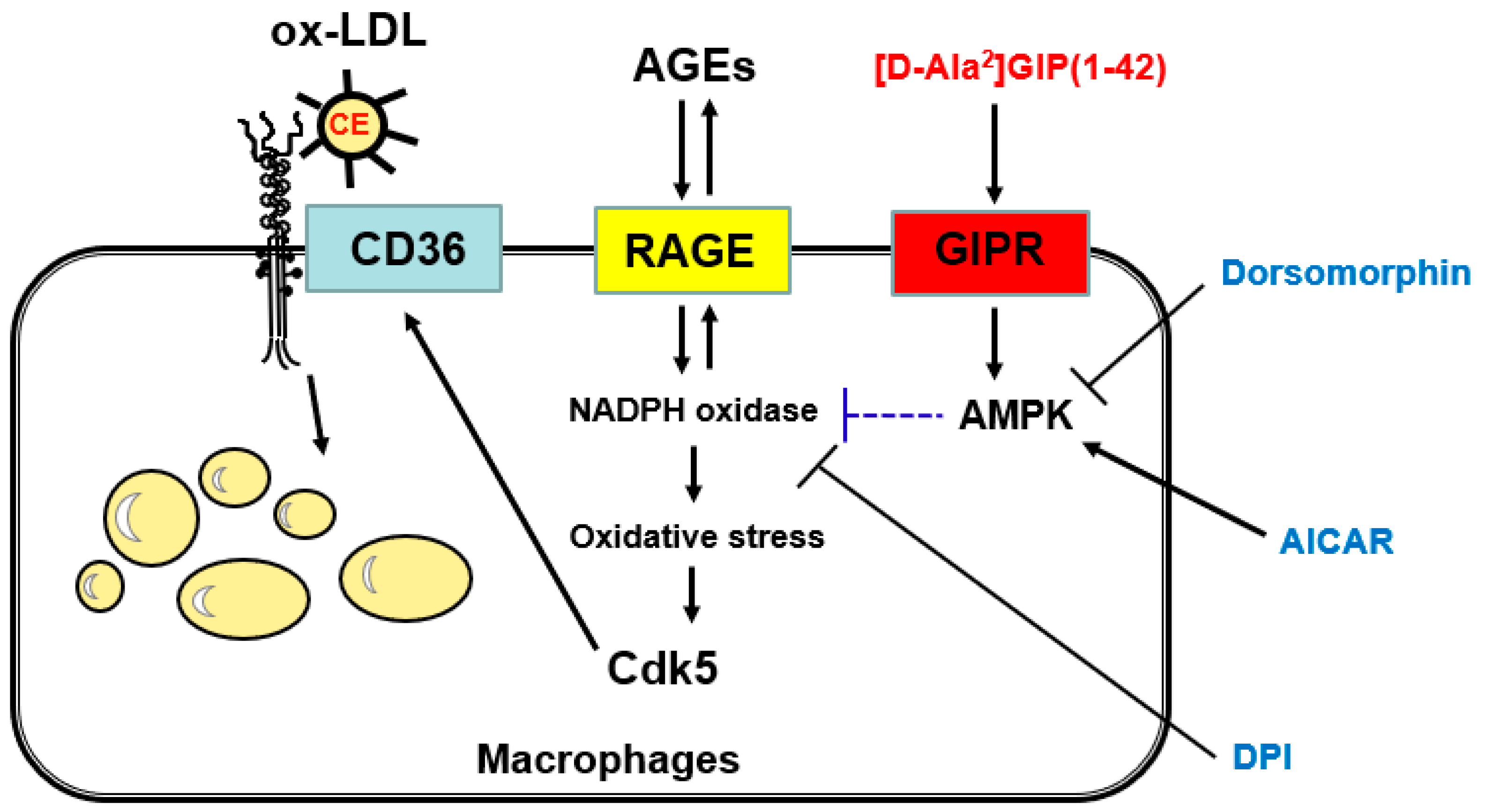
3. Discussion
4. Materials and Methods
4.1. Chemical Regents and Materials
4.2. Preparation of AGEs of Bovine Serum Albumin (BSA)
4.3. Cell Culture of U937 Monocyte-Derived Macrophages
4.4. Measurement of Fluorescence Intensity of Dil-ox-LDL and ROS Generation in U937 Cells
4.5. Quantitative Real-Time Reverse Transcription–Polymerase Chain Reaction (RT-PCR)
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef]
- Fukami, A.; Seino, Y.; Ozaki, N.; Yamamoto, M.; Sugiyama, C.; Sakamoto-Miura, E.; Himeno, T.; Takagishi, Y.; Tsunekawa, S.; Ali, S.; et al. Ectopic expression of GIP in pancreatic β-cells maintains enhanced insulin secretion in mice with complete absence of proglucagon-derived peptides. Diabetes 2013, 62, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef]
- Seino, Y.; Yabe, D. Glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1: Incretin actions beyond the pancreas. J. Diabetes Investig. 2013, 4, 108–130. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Ahrén, B. G-protein-coupled receptors and islet function-implications for treatment of type 2 diabetes. Pharmacol. Ther. 2007, 116, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Siegel, E.G.; Creutzfeldt, W. Stimulation of insulin release in isolated rat islets by GIP in physiological concentrations and its relation to islet cyclic AMP content. Diabetologia 1985, 28, 857–861. [Google Scholar] [CrossRef]
- Coskun, T.; Sloop, K.W.; Loghin, C.; Alsina-Fernandez, J.; Urva, S.; Bokvist, K.B.; Cui, X.; Briere, D.A.; Cabrera, O.; Roell, W.C.; et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol. Metab. 2018, 18, 3–14. [Google Scholar] [CrossRef]
- Inagaki, N.; Takeuchi, M.; Oura, T.; Imaoka, T.; Seino, Y. Efficacy and safety of tirzepatide monotherapy compared with dulaglutide in Japanese patients with type 2 diabetes (SURPASS J-mono): A double-blind, multicentre, randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2022, 10, 623–633. [Google Scholar] [CrossRef]
- Huang, X.; Liu, J.; Peng, G.; Lu, M.; Zhou, Z.; Jiang, N.; Yan, Z. Gut hormone multi-agonists for the treatment of type 2 diabetes and obesity: Advances and challenges. J. Endocrinol. 2024, 262, e230404. [Google Scholar] [CrossRef] [PubMed]
- Usdin, T.B.; Mezey, E.; Button, D.C.; Brownstein, M.J.; Bonner, T.I. Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 1993, 133, 2861–2870. [Google Scholar] [CrossRef]
- Drucker, D.J.; Holst, J.J. The expanding incretin universe: From basic biology to clinical translation. Diabetologia 2023, 66, 1765–1779. [Google Scholar] [CrossRef] [PubMed]
- Nogi, Y.; Nagashima, M.; Terasaki, M.; Nohtomi, K.; Watanabe, T.; Hirano, T. Glucose-dependent insulinotropic polypeptide prevents the progression of macrophage-driven atherosclerosis in diabetic apolipoprotein E-null mice. PLoS ONE 2012, 7, e35683. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Kushima, H.; Koshibu, M.; Saito, T.; Hiromura, M.; Kohashi, K.; Terasaki, M.; Seino, Y.; Yamada, Y.; Hirano, T. Glucose-dependent insulinotropic polypeptide suppresses peripheral arterial remodeling in male mice. Endocrinology 2018, 159, 2717–2732. [Google Scholar] [CrossRef]
- Ojima, A.; Matsui, T.; Maeda, S.; Takeuchi, M.; Yamagishi, S. Glucose-dependent insulinotropic polypeptide (GIP) inhibits signaling pathways of advanced glycation end products (AGEs) in endothelial cells via its antioxidative properties. Horm. Metab. Res. 2012, 44, 501–505. [Google Scholar] [CrossRef]
- Kahles, F.; Liberman, A.; Halim, C.; Rau, M.; Möllmann, J.; Mertens, R.W.; Rückbeil, M.; Diepolder, I.; Walla, B.; Diebold, S.; et al. The incretin hormone GIP is upregulated in patients with atherosclerosis and stabilizes plaques in ApoE−/− mice by blocking monocyte/macrophage activation. Mol. Metab. 2018, 14, 150–157. [Google Scholar] [CrossRef]
- Terasaki, M.; Yashima, H.; Mori, Y.; Saito, T.; Shigara, Y.; Kawakami, R.; Ohara, M.; Fukui, T.; Hirano, T.; Yamada, Y.; et al. Glucose-dependent insulinotropic polypeptide suppresses foam cell formation of macrophages through inhibition of the cyclin-dependent kinase 5-CD36 pathway. Biomedicines 2021, 9, 832. [Google Scholar] [CrossRef]
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar]
- Damaskos, C.; Garmpis, N.; Kollia, P.; Mitsiopoulos, G.; Barlampa, D.; Drosos, A.; Patsouras, A.; Gravvanis, N.; Antoniou, V.; Litos, A.; et al. Assessing Cardiovascular Risk in Patients with Diabetes: An Update. Curr. Cardiol. Rev. 2020, 16, 266–274. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I. Role of advanced glycation endproduct (AGE)-receptor for advanced glycation endproduct (RAGE) axis in cardiovascular disease and its therapeutic intervention. Circ. J. 2019, 83, 1822–1828. [Google Scholar] [CrossRef]
- Razaei, M.; Rabizadeh, S.; Mirahmad, M.; Hajmiri, M.S.; Nakhjavani, M.; Hemmatabadi, M.; Shirzad, N. The association between advanced glycation end products (AGEs) and ABC (hemoglobin A1C, blood pressure, and low-density lipoprotein cholesterol) control parameters among patients with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2022, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Jud, P.; Sourij, H. Therapeutic options to reduce advanced glycation end products in patients with diabetes mellitus: A review. Diabetes Res. Clin. Pract. 2019, 148, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Kilhovd, B.K.; Juutilainen, A.; Lehto, S.; Rönnemaa, T.; Torjesen, P.A.; Hanssen, K.F.; Laakso, M. Increased serum levels of methylglyoxal-derived hydroimidazolone-AGE are associated with increased cardiovascular disease mortality in nondiabetic women. Atherosclerosis 2009, 205, 590–594. [Google Scholar] [CrossRef]
- Kilhovd, B.K.; Juutilainen, A.; Lehto, S.; Rönnemaa, T.; Torjesen, P.A.; Hanssen, K.F.; Laakso, M. Increased serum levels of advanced glycation endproducts predict total, cardiovascular and coronary mortality in women with type 2 diabetes: A population-based 18 year follow-up study. Diabetologia 2007, 50, 1409–1417. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Stern, D. Atherosclerosis and diabetes: The RAGE connection. Curr. Atheroscler. Rep. 2000, 2, 430–436. [Google Scholar] [CrossRef]
- Bijnen, M.; Beelen, N.; Wetzels, S.; Gaar, J.V.; Vroomen, M.; Wijnands, E.; Scheijen, J.L.; van de Waarenburg, M.P.H.; Gijbels, M.J.; Cleutjens, J.P.; et al. RAGE deficiency dose not affect non-alcholic steatohepatitis and atherosclerosis in Western type diet-fed Ldlr(−/−) mice. Sci. Rep. 2018, 8, 15256. [Google Scholar] [CrossRef]
- Han, X.; Ma, W.; Zhu, Y.; Sun, X.; Liu, N. Advanced glycation end products enhance macrophage polarization to the Ma phenotype via the HIF-1α/PDK4 pathway. Mol. Cell. Endocrinol. 2020, 514, 110878. [Google Scholar] [CrossRef]
- Hassen, N.M.; Wouters, K.; Hujiberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a repture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Jing, L.L.; Yan, J.C.; Sun, Z.; Bao, Z.Y.; Shao, C.; Pang, Q.W.; Geng, Y.; Zhang, L.L.; Li, L.H. Role of AGEs in the progression and regression of atherosclerotic plaques. Glycoconj. J. 2018, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Takeya, M.; Mori, T.; Araki, N.; Suzuki, H.; Horiuchi, S.; Kodama, T.; Miyauchi, Y.; Takahashi, K. Immunohistochemical and ultrastructural detection of advanced glycation end products in atherosclerotic lesions of human aorta with a novel specific monoclonal antibody. Am. J. Pathol. 1995, 147, 654–667. [Google Scholar] [PubMed]
- Nin, J.W.; Jorsal, A.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D. Higher plasma soluble receptor for advanced glycation end products (sRAGE) levels are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: A 12-year follow-up study. Diabetes 2010, 59, 2027–2032. [Google Scholar] [CrossRef]
- Nin, J.W.; Jorsal, A.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D. Higher plasma levels of advanced glycation end products are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: A 12-year follow-up study. Diabetes Care 2011, 34, 442–447. [Google Scholar] [CrossRef]
- Semba, R.D.; Bandineli, S.; Sun, K.; Guralnik, J.M.; Ferrucci, L. Plasma carboxymethyl-lysine, an advanced glycation end product, and all-cause and cardiovascular disease mortality in older community-dwelling adults. J. Am. Geriatr. Soc. 2009, 57, 1874–1880. [Google Scholar] [CrossRef]
- Van Waateringe, R.P.; Fokkens, B.T.; Slagter, S.N.; van der Klauw, M.M.; van Vliet-Ostaptchouk, J.V.; Graaff, R.; Paterson, A.D.; Smit, A.J.; Lutgers, H.L.; Wolffenbuttel, B.H.R. Skin autofluorescence predicts incident type 2 diabetes, cardiovascular disease and mortality in the general population. Diabetologia 2019, 62, 269–280. [Google Scholar] [CrossRef]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S. RAGE-aptamer blocks the development and progression of experimental diabetic nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef]
- Terasaki, M.; Yashima, H.; Mori, Y.; Saito, T.; Matsui, T.; Hiromura, M.; Kushima, H.; Osaka, N.; Ohara, M.; Fukui, T.; et al. A dipeptidyl peptidase-4 inhibitor inhibits foam cell formation of macrophages in type 1 diabetes via suppression of CD36 expression. Int. J. Mol. Sci. 2020, 21, 4811. [Google Scholar] [CrossRef]
- Yashima, H.; Terasaki, M.; Sotokawauchi, A.; Matsui, T.; Mori, Y.; Saito, T.; Osaka, N.; Kushima, H.; Hiromura, M.; Ohara, M.; et al. AGE-RAGE axis stimulates oxidized LDL uptake into macrophages through cyclin-dependent kinase 5-CD36 pathway via oxidative stress generation. Int. J. Mol. Sci. 2020, 21, 9263. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.; Lewis, J.; Rao, H.; Carter, J.; Portillo, I.; Beuttler, R. Effects of GLP-1 receptor agonists on cardiovascular outcomes in patients with type 2 diabetes and chronic kidney disease: A systematic review and meta-analysis. Pharmacotherapy 2022, 42, 921–928. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef]
- Wen, S.Y.; Zhi, X.; Liu, H.X.; Wang, X.; Chen, Y.Y.; Wang, L. Is the suppression of CD36 a promising way for atherosclerosis therapy? Biochem. Pharmacol. 2024, 219, 115965. [Google Scholar] [CrossRef]
- Duan, H.; Song, P.; Li, R.; Su, H.; He, L. Attenuating lipid metabolism in atherosclerosis: The potential role of Anti-oxidative effects on low-density lipoprotein of herbal medicines. Front. Pharmacol. 2023, 14, 1161657. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species mediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef] [PubMed]
- Ping, M.; Xiao, W.; Mo, L.; Xiao, X.; Song, S.; Tang, W.; Yang, X. Paeonol attenuates advanced oxidation protein product-induced oxidative stress injury in THP-1 macrophages. Pharmacology 2014, 93, 286–295. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Takeuchi, M.; Yamagishi, S. Metformin inhibits advanced glycation end products (AGEs)-induced growth and VEGF expression in MCF-7 breast cancer cells by suppressing AGEs receptor expression via AMP-activated protein kinase. Horm. Metab. Res. 2013, 45, 387–390. [Google Scholar] [CrossRef]
- Nassif, R.M.; Chalhoub, E.; Chedid, P.; Hurtado-Nedelec, M.; Raya, E.; Dang, P.M.; Marie, J.C.; El-Benna, J. Metformin Inhibits ROS Production by Human M2 Macrophages via the Activation of AMPK. Biomedicines 2022, 10, 319. [Google Scholar] [CrossRef]
- Kim, M.J.; Nagy, L.E.; Park, P.H. Globular adiponectin inhibits ethanol-induced reactive oxygen species production through modulation of NADPH oxidase in macrophages: Involvement of liver kinase B1/AMP-activated protein kinase pathway. Mol. Pharmacol. 2014, 86, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Ewart, M.A.; Kennedy, S. AMPK and vasculoprotection. Pharmacol. Ther. 2011, 131, 242–253. [Google Scholar] [CrossRef]
- Gao, F.; Chen, J.; Zhu, H. A potential strategy for treating atherosclerosis: Improving endothelial function via AMP-activated protein kinase. Sci. China Life Sci. 2018, 61, 1024–1029. [Google Scholar] [CrossRef]
- Carling, D.; Sanders, M.J.; Woods, A. The regulation of AMP-activated protein kinase by upstream kinases. Int. J. Obes. 2008, 32 (Suppl. S4), S55–S59. [Google Scholar] [CrossRef]
- Bourdonnay, E.; Serezani, C.H.; Aronoff, D.M.; Peters-Golden, M. Regulation of alveolar macrophage p40phox: Hierarchy of activating kinases and their inhibition by PGE2. J. Leukoc. Biol. 2012, 92, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Tomas, A.; Rutter, G.A. Effects on pancreatic Beta and other Islet cells of the glucose-dependent insulinotropic polypeptide. Peptides 2020, 125, 170201. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Ramos, L.S.; Buck, J.; Levin, L.R.; Rubino, F.; McGraw, T.E. Gastric inhibitory peptide controls adipose insulin sensitivity via activation of cAMP-response element-binding protein and p110β isoform of phosphatidylinositol 3-kinase. J. Biol. Chem. 2011, 286, 43062–43070. [Google Scholar] [CrossRef]
- Terasaki, M.; Shibata, K.; Mori, Y.; Saito, T.; Matsui, T.; Ohara, M.; Fukui, T.; Hasumi, K.; Higashimoto, Y.; Nobe, K.; et al. SMTP-44D inhibits atherosclerotic plaque formation in apolipoprotein-E null mice partly by suppressing the AGEs-RAGE axis. Int. J. Mol. Sci. 2023, 24, 6505. [Google Scholar] [CrossRef]
- Huh, H.Y.; Pearce, S.F.; Yesner, L.M.; Schindler, J.L.; Silerstein, R.L. Regulated expression of CD36 during monocyte-to-macrophage differentiation: Potential role of CD36 in foam cell formation. Blood 1996, 87, 2020–2028. [Google Scholar] [CrossRef]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Bostrom, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Bluher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef]
- Leonarduzzi, G.; Gamba, P.; Gargiulo, S.; Sottero, B.; Kadl, A.; Biasi, F.; Chiarpotto, E.; Leitinger, N.; Vendemiale, G.; Serviddio, G.; et al. Oxidation as a crucial reaction for cholesterol to induce tissue degeneration: CD36 overexpression in human promonocytic cells treated with a biologically relevant oxysterol mixture. Aging Cell 2008, 7, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Makita, Z.; Yanagisawa, K.; Kameda, K.; Koike, T. Detection of noncarboxymethyllysine and carboxymethyllysine advanced glycation end products (AGE) in serum of diabetic patients. Mol. Med. 1999, 5, 393–405. [Google Scholar] [CrossRef]
- Sylvester, A.L.; Zhang, D.X.; Ran, S.; Zinkevich, N.S. Inhibiting NADPH Oxidases to Target Vascular and Other Pathologies: An Update on Recent Experimental and Clinical Studies. Biomolecules 2022, 12, 823. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T. Pigment Epithelium-Derived Factor: A Novel Therapeutic Target for Cardiometabolic Diseases and Related Complications. Curr. Med. Chem. 2018, 25, 1480–1500. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Y.; Zhang, X.; Xu, Z.; Zhou, J.; Shang, W. SPP-4 inhibitor linagliptin ameliorates oxidized LDL-induced THP-1 macrophage foam cell formation and inflammation. Drug Des. Dev. Ther. 2020, 14, 3929–3940. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Xu, Q.; Xu, C.; Shi, W.; Pang, R.; Zhang, K.; Liang, X.; Li, H.; Li, Z.; et al. Dexamethasone induced osteocyte apoptosis in steroid-induced femoral head osteonecrosis through ROS-mediated oxidative stress. Orthop. Surg. 2024, 16, 733–744. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terasaki, M.; Yashima, H.; Mori, Y.; Saito, T.; Inoue, N.; Matsui, T.; Osaka, N.; Fujikawa, T.; Ohara, M.; Yamagishi, S.-i. Glucose-Dependent Insulinotropic Polypeptide Inhibits AGE-Induced NADPH Oxidase-Derived Oxidative Stress Generation and Foam Cell Formation in Macrophages Partly via AMPK Activation. Int. J. Mol. Sci. 2024, 25, 9724. https://doi.org/10.3390/ijms25179724
Terasaki M, Yashima H, Mori Y, Saito T, Inoue N, Matsui T, Osaka N, Fujikawa T, Ohara M, Yamagishi S-i. Glucose-Dependent Insulinotropic Polypeptide Inhibits AGE-Induced NADPH Oxidase-Derived Oxidative Stress Generation and Foam Cell Formation in Macrophages Partly via AMPK Activation. International Journal of Molecular Sciences. 2024; 25(17):9724. https://doi.org/10.3390/ijms25179724
Chicago/Turabian StyleTerasaki, Michishige, Hironori Yashima, Yusaku Mori, Tomomi Saito, Naoto Inoue, Takanori Matsui, Naoya Osaka, Tomoki Fujikawa, Makoto Ohara, and Sho-ichi Yamagishi. 2024. "Glucose-Dependent Insulinotropic Polypeptide Inhibits AGE-Induced NADPH Oxidase-Derived Oxidative Stress Generation and Foam Cell Formation in Macrophages Partly via AMPK Activation" International Journal of Molecular Sciences 25, no. 17: 9724. https://doi.org/10.3390/ijms25179724