
Efficient Solution-Phase Dipeptide Synthesis Using Titanium Tetrachloride and Microwave Heating


Abstract
1. Introduction
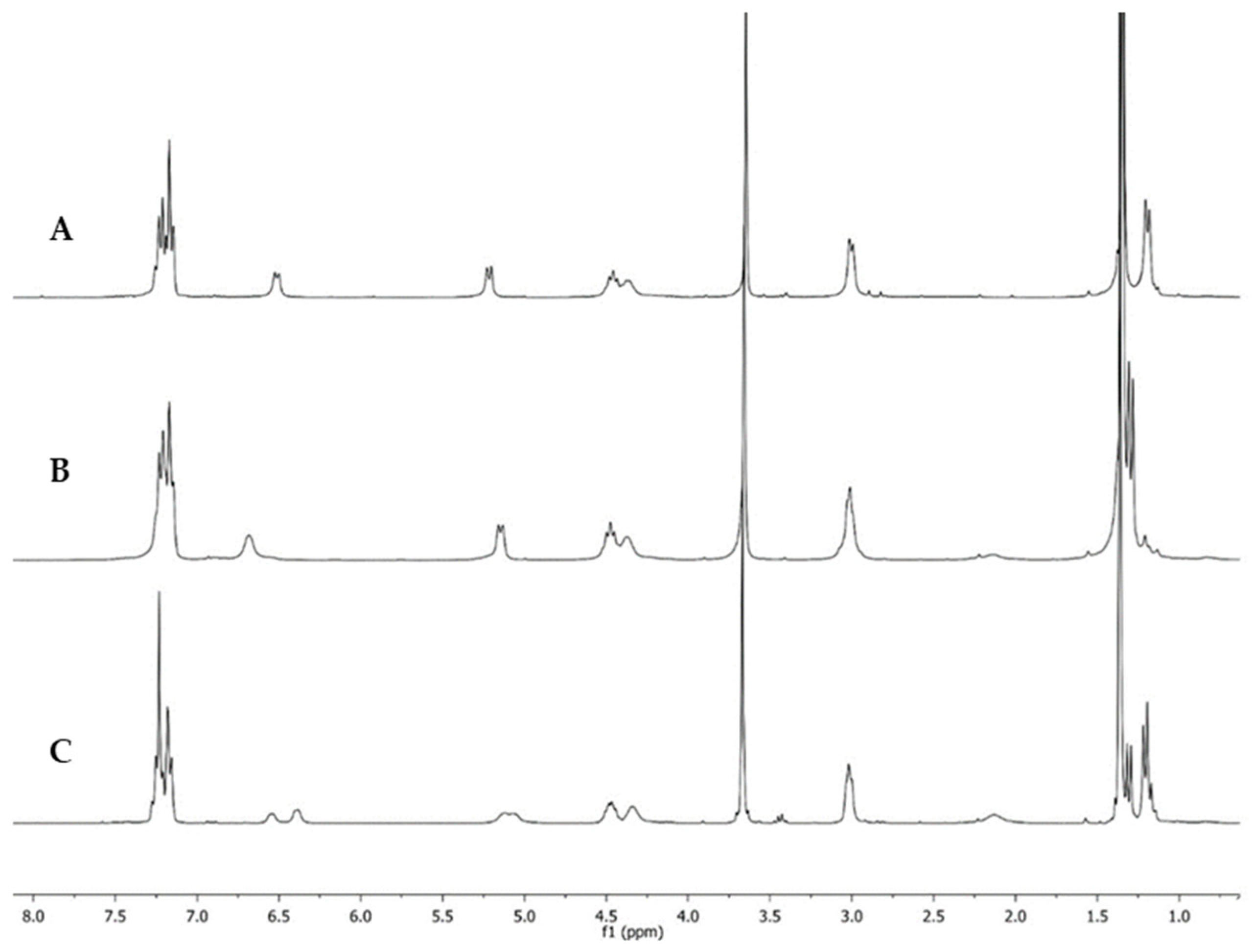
2. Results and Discussion
3. Materials and Methods
3.1. General Information
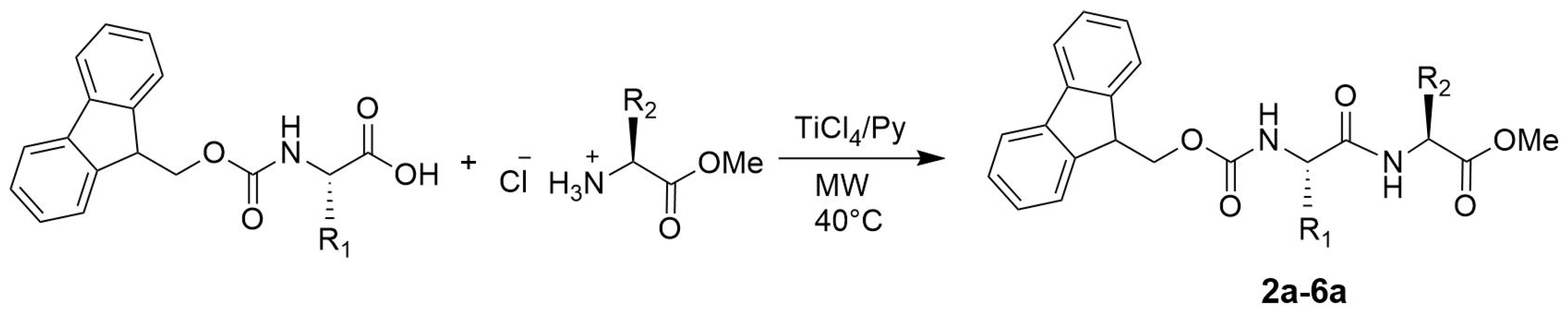
3.2. General Procedure for the Synthesis of N-Protected Dipeptides
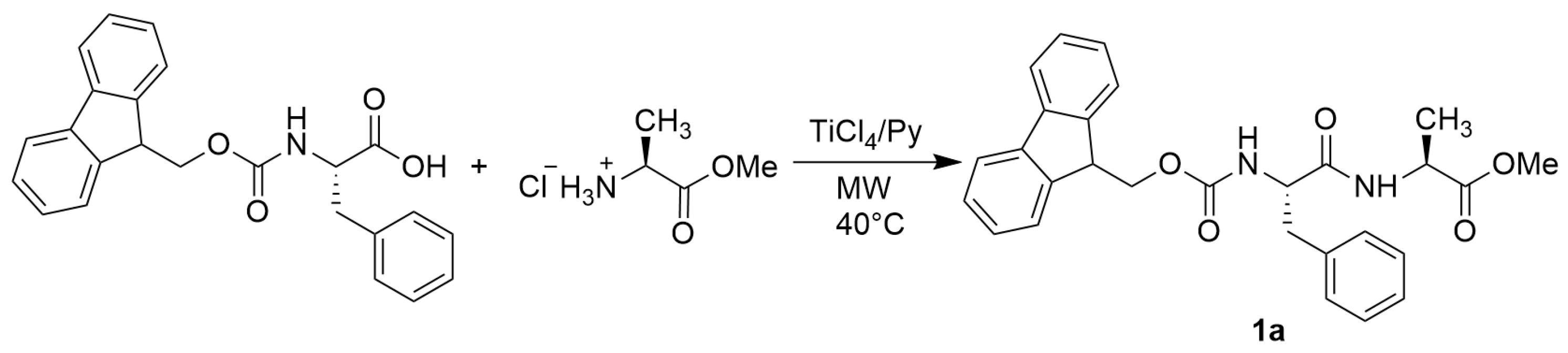
- N-Fmoc-L-Phe-L-Ala-OMe (1a)
- N-Fmoc-Gly-L-Ala-OMe (2a)
- N-Fmoc-L-Tyr(tBu)-Gly-OMe (3a)
- N-Fmoc-L-Leu-Gly-OMe (4a)
- N-Fmoc-L-Leu-L-Ile-OMe (5a)
- N-Fmoc-L-Cys(Bzl)-L-Ala-OMe (6a)
- N-Boc-L-Phe-L-Ala-OMe (1b)
- N-Boc-L-Phe-D-Ala-OMe (2b)
- N-Boc-D-Phe-D-Ala-OMe (3b)
- N-Boc-L-Ile-L-Leu-OMe (4b)
- N-Boc-L-Leu-L-Val-OMe (5b)
- N-Boc-L-Arg(Z)2-L-Ala-OMe (6b)
- N-Boc-L-Phe-L-Leu-OMe (7b)
- N-Z-L-Ala-L-Ala-OMe (1c)
- N-Z-Gly-L-Val-OMe (2c)
- N-Z-L-Val-L-Phe-OMe (3c)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance, Chemistry, Biochemistry and Material Science; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Kaspar, A.A.; Reichert, J.M. Future directions for peptide therapeutics development. Drug Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Thorner, J.; Emr, S.D.; Abelson, J.N. Applications of chimeric genes and hybrid proteins part A: Gene expression and protein purification. In Methods Enzymology, 1st ed.; Academic Press: Cambridge, MA, USA, 2000; Volume 326, pp. 601–617. [Google Scholar]
- Forbes, J.; Krishnamurthy, K. Biochemistry, Peptide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK562260 (accessed on 28 August 2024).
- Rossino, G.; Marchese, E.; Galli, G.; Verde, F.; Finizio, M.; Serra, M.; Linciano, P.; Collina, S. Peptides as Therapeutic Agents: Challenges and Opportunities in the Green Transition Era. Molecules 2023, 28, 7165. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Nicolás-Morales, M.L.; Luisa-Sanjuan, A.; Gutiérrez-Torres, M.; Vences-Velázquez, A.; Ortuño-Pineda, C.; Espinoza-Rojo, M.; Navarro-Tito, N.; Cortés-Sarabia, K. Peptide-Based Vaccines in Clinical Phases and New Potential Therapeutic Targets as a New Approach for Breast Cancer: A Review. Vaccines 2022, 10, 1249. [Google Scholar] [CrossRef] [PubMed]
- Vadevoo, S.M.P.; Gurung, S.; Lee, H.S.; Gunassekaran, G.R.; Lee, S.M.; Yoon, J.W.; Lee, Y.K.; Lee, B. Peptides as multifunctional players in cancer therapy. Exp. Mol. Med. 2023, 55, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Pressley, H.; Cornelio, C.K.; Adams, E.N. Setmelanotide: A Novel Targeted Treatment for Monogenic Obesity. J. Pharm. Technol. 2022, 38, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Trofinetide: First Approval. Drugs 2023, 83, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, T.; Lerma, E.; Engelke, K.; Huizinga, R.B. Voclosporin: A novel calcineurin inhibitor for the treatment of lupus nephritis. Expert. Rev. Clin. Pharmacol. 2022, 15, 515–529. [Google Scholar] [CrossRef]
- Keam, S.J. Lutetium Lu 177 Vipivotide Tetraxetan: First Approval. Mol. Diagn. Ther. 2022, 26, 467–475. [Google Scholar] [CrossRef]
- Sharma, K.; Sharma, K.K.; Sharma, A.; Jain, R. Peptide-based drug discovery: Current status and recent advances. Drug Discov. Today 2023, 28, 103464. [Google Scholar] [CrossRef]
- Acosta-Guzmán, P.; Ojeda-Porras, A.; Gamba-Sánchez, D. Contemporary approaches for amide bond formation. Adv. Synth. Catal. 2023, 365, 4359. [Google Scholar] [CrossRef]
- de Figueiredo, R.M.; Suppo, J.S.; Campagne, J.M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Spinella, M.; De Lorenzo, A.; Leggio, A.; Liguori, A. C → N and N → C solution phase peptide synthesis using the N-acyl 4-nitrobenzenesulfonamide as protection of the carboxylic function. Org. Biomol. Chem. 2013, 11, 3786–3796. [Google Scholar] [CrossRef] [PubMed]
- Leggio, A.; Belsito, E.L.; De Luca, G.; Di Gioia, M.L.; Leotta, V.; Romio, E.; Siciliano, C.; Liguori, A. One-pot synthesis of amides from carboxylic acids activated using thionyl chloride. RSC Adv. 2016, 6, 34468–34475. [Google Scholar] [CrossRef]
- Leggio, A.; Comandè, A.; Belsito, E.L.; Greco, M.; Lo Feudo, L.; Liguori, A. Alternative formation of amides and β-enaminones from aroyl chlorides using the TiCl4-trialkylamine reagent system. Org. Biomol. Chem. 2018, 16, 5677–5683. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Szostak, M. Non-Classical Amide Bond Formation: Transamidation and Amidation of Activated Amides and Esters by Selective N–C/O–C Cleavage. Synthesis 2020, 52, 2579–2599. [Google Scholar]
- Mijalis, A.J.; Thomas, D.A., 3rd; Simon, M.D.; Adamo, A.; Beaumont, R.; Jensen, K.F.; Pentelute, B.L. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol. 2017, 13, 464–466. [Google Scholar] [CrossRef]
- Naoum, J.N.; Alshanski, I.; Mayer, G.; Strauss, P.; Hurevich, M. Stirring Peptide Synthesis to a New Level of Efficiency. Org. Process Res. Dev. 2022, 26, 129–136. [Google Scholar] [CrossRef]
- Sanchez-Sancho, F.; Mann, E.; Herradon, B. Efficient syntheses of polyannular heterocycles featuring microwave-accelerated Bischler-Napieralski reaction, stereoselective Heck cyclization, and Claisen rearrangement. Synlett 2000, 2000, 509–513. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide Coupling Reagents, More than a Letter Soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar]
- Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A preparation of N-Fmoc-N-methyl-α-amino acids and N-nosyl-N-methyl-α-amino acids. Amino Acids 2010, 38, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; Tu, Z.; McElveen, E.; Xu, J.; Taylor, M.; Luedtke, R.R.; Mach, R.H. Synthesis and in vitro binding of N-phenyl piperazine analogs as potential dopamine D3 receptor ligands. Bioorg. Med. Chem. 2005, 13, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Gacesa, R.; Tripodi, A.A.P.; Cilibrizzi, A.; Leggio, A.; Hider, R.; Abbate, V. Solid-Phase Synthesis and In-Silico Analysis of Iron-Binding Catecholato Chelators. Int. J. Mol. Sci. 2020, 21, 7498. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.C.; Trejo-Martin, A.; Chilton, M.L.; Kostal, J.; Bercu, J.; Beutner, G.L.; Bruen, U.S.; Dolan, D.G.; Gomez, S.; Hillegass, J.; et al. An Evaluation of the Occupational Health Hazards of Peptide Couplers. Chem. Res. Toxicol. 2022, 35, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Xu, J.-C. 1-Ethyl 2-halopyridinium salts, highly efficient coupling reagents for hindered peptide synthesis both in solution and the solid-phase. Tetrahedron 2000, 56, 8119–8131. [Google Scholar]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [PubMed]
- Prabhu, G.; Narendra, N.; Vishwanatha, T.M.; Sureshbabu, V.V. Amino acid chlorides: A journey from instability and racemization toward broader utility in organic synthesis including peptides and their mimetics. Tetrahedron 2015, 71, 2785–2832. [Google Scholar] [CrossRef]
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014, 43, 2714–2742. [Google Scholar] [CrossRef]
- Lundberg, H.; Tinnis, F.; Adolfsson, H. Direct amide coupling of non-activated carboxylic acids and amines catalysed by zirconium(IV) chloride. Chemistry 2012, 18, 3822–3826. [Google Scholar] [CrossRef]
- Muramatsu, W.; Hattori, T.; Yamamoto, H. Amide bond formation: Beyond the dilemma between activation and racemisation. Chem. Commun. 2021, 57, 6346–6359. [Google Scholar] [CrossRef]
- Muramatsu, W.; Hattori, T.; Yamamoto, H.J. Substrate-directed lewis-acid catalysis for peptide synthesis. J. Am. Chem. Soc. 2019, 141, 12288–12295. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, H.; Tinnis, F.; Adolfsson, H. Titanium (IV) Isopropoxide as an Efficient Catalyst for Direct Amidation of Nonactivated Carboxylic Acids. Synlett 2012, 23, 2201–2204. [Google Scholar] [CrossRef]
- Todorovic, M.; Perrin, D.M. Recent developments in catalytic amide bond formation. Pept. Sci. 2020, 112, e24210. [Google Scholar] [CrossRef]
- Nordahl, Å.; Carlson, R. Carboxamides from Carboxylic Acids by Lewis Acid Catalysis. Use of Principal Properties for Exploring Different Reaction Conditions. Acta Chem. Scand., Ser. B 1988, 42, 28–34. [Google Scholar] [CrossRef]
- Shteinberg, L.Y.; Kondratov, S.A.; Shein, S.M. Metal-complex catalysis under aniline acylation by substituted benzoic acids. J. Org. Khim. 1988, 24, 1968–1972. [Google Scholar]
- Di Gioia, M.L.; Leggio, A.; Guarino, I.F.; Leotta, V.; Romio, E.; Liguori, A. A simple synthesis of anilines by LiAlH4/TiCl4 reduction of aromatic nitro compounds. Tetrahedron Lett. 2015, 56, 5341–5344. [Google Scholar] [CrossRef]
- Cavallaro, P.A.; De Santo, M.; Greco, M.; Marinaro, R.; Belsito, E.L.; Liguori, A.; Leggio, A. Titanium Tetrachloride-Assisted Direct Esterification of Carboxylic Acids. Molecules 2024, 29, 777. [Google Scholar] [CrossRef]
- Leggio, A.; Bagalà, J.; Belsito, E.L.; Comandè, A.; Greco, M.; Liguori, A. Formation of amides: One-pot condensation of carboxylic acids and amines mediated by TiCl4. Chem. Cent. J. 2017, 11, 87. [Google Scholar] [CrossRef]
- Comandè, A.; Greco, M.; Belsito, E.L.; Liguori, A.; Leggio, A. A titanium tetrachloride-based effective methodology for the synthesis of dipeptides. RSC Adv. 2019, 9, 22137–22142. [Google Scholar] [CrossRef]
- De la Hoz, A.; Dìaz-Ortiz, A.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. Engl. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed]
- Adam, D. Out of the kitchen. Nature 2003, 421, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.M.; Leadbeater, N.E. Microwave energy: A versatile tool for the biosciences. Org. Biomol. Chem. 2007, 5, 1141–1150. [Google Scholar] [CrossRef]
- Herrero, M.A.; Kremsner, J.M.; Kappe, C.O. Nonthermal Microwave Effects Revisited: On the Importance of Internal Temperature Monitoring and Agitation in Microwave Chemistry. J. Org. Chem. 2008, 73, 36–47. [Google Scholar] [CrossRef]
- Hoz, A.; Loupy, A. Microwaves in Organic Synthesis, 3rd ed.; Wiley: Weinheim, Germany, 2012. [Google Scholar]
- Mahindra, A.; Sharma, K.K.; Jain, R. Rapid microwave-assisted solution-phase peptide synthesis. Tetrahedron Lett. 2012, 53, 6931–6935. [Google Scholar] [CrossRef]
- Mahindra, A.; Nooney, K.; Uraon, S.; Sharma, K.K.; Jain, R. Microwave-assisted solution phase peptide synthesis in neat water. RSC Adv. 2013, 3, 16810–16816. [Google Scholar] [CrossRef]
- Priecel, P.; Lopez-Sanchez, J.A. Advantages and Limitations of Microwave Reactors: From Chemical Synthesis to the Catalytic Valorization of Biobased Chemicals. ACS Sustain. Chem. Eng. 2019, 7, 3–21. [Google Scholar] [CrossRef]
- Tosi, E.; Campagne, J.-M.; Marcia de Figueiredo, R. Amine Activation: “Inverse” Dipeptide Synthesis and Amide Function Formation through Activated Amino Compounds. J. Org. Chem. 2022, 87, 12148–12163. [Google Scholar] [CrossRef]
- Liu, D.; Guo, Y.-L.; Qu, J.; Zhang, C. Recyclable hypervalent-iodine-mediated solid-phase peptide synthesis and cyclic peptide synthesis. Beilstein J. Org. Chem. 2018, 14, 1112–1119. [Google Scholar]
- Chen, H.; Xu, X.; Liu, L.; Tang, G.; Zhao, Y. Phosphorus oxychloride as an efficient coupling reagent for the synthesis of esters, amides and peptides under mild conditions. RSC Adv. 2013, 3, 16247–16250. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dipeptide | Temperature °C | Power (Watt) | Time (min) | Yield a (%) |
|---|---|---|---|---|
| 1a | 40 °C | 50 | 60 | 53 |
| 1a | 40 °C | 100 | 40 | 70 |
| 1a | 40 °C | 250 | 20 | 90 |
| Dipeptide | R1 | R2 | Time (min) | Yield a (%) |
|---|---|---|---|---|
| 2a | H | CH3 | 30 | 88 |
| 3a | CH2C6H4OtBu | H | 25 | 78 |
| 4a | CH2CH(CH3)2 | H | 20 | 80 |
| 5a | CH2CH(CH3)2 | CH(CH3)CH2CH3 | 40 | 70 |
| 6a | CH2SCH2C6H5 | CH3 | 35 | 70 |
| Dipeptide | R1 | R2 | Time (min) | Yield a (%) |
|---|---|---|---|---|
| 1b | CH2C6H5 (L) | CH3 (L) | 20 | 94 |
| 2b | CH2C6H5 (L) | CH3 (D) | 20 | 76 |
| 3b | CH2C6H5 (D) | CH3 (D) | 25 | 71 |
| 4b | CH(CH3)CH2CH3 | CH2CH(CH3)2 | 35 | 84 |
| 5b | CH2CH(CH3)2 | CH(CH3)2 | 30 | 70 |
| 6b | (CH2)3NHCN(Z)NH(Z) | CH3 | 40 | 83 |
| 7b | CH2C6H5 | CH2CH(CH3)2 | 25 | 89 |
| Dipeptide | R1 | R2 | Time (min) | Yield a (%) |
|---|---|---|---|---|
| 1c | CH2C6H5 | CH3 | 30 | 81 |
| 2c | H | CH(CH3)2 | 30 | 75 |
| 3c | CH(CH3)2 | CH2C6H5 | 35 | 65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavallaro, P.A.; De Santo, M.; Marinaro, R.; Belsito, E.L.; Liguori, A.; Leggio, A. Efficient Solution-Phase Dipeptide Synthesis Using Titanium Tetrachloride and Microwave Heating. Int. J. Mol. Sci. 2024, 25, 9729. https://doi.org/10.3390/ijms25179729
Cavallaro PA, De Santo M, Marinaro R, Belsito EL, Liguori A, Leggio A. Efficient Solution-Phase Dipeptide Synthesis Using Titanium Tetrachloride and Microwave Heating. International Journal of Molecular Sciences. 2024; 25(17):9729. https://doi.org/10.3390/ijms25179729
Chicago/Turabian StyleCavallaro, Palmira Alessia, Marzia De Santo, Rocco Marinaro, Emilia Lucia Belsito, Angelo Liguori, and Antonella Leggio. 2024. "Efficient Solution-Phase Dipeptide Synthesis Using Titanium Tetrachloride and Microwave Heating" International Journal of Molecular Sciences 25, no. 17: 9729. https://doi.org/10.3390/ijms25179729
APA StyleCavallaro, P. A., De Santo, M., Marinaro, R., Belsito, E. L., Liguori, A., & Leggio, A. (2024). Efficient Solution-Phase Dipeptide Synthesis Using Titanium Tetrachloride and Microwave Heating. International Journal of Molecular Sciences, 25(17), 9729. https://doi.org/10.3390/ijms25179729