
Gene Therapy for Achromatopsia


Abstract
:1. Introduction
2. Achromatopsia Phenotypic Patterns
3. Genetic Basis of Achromatopsia Management
4. Diagnosis and Testing for Achromatopsia
5. Achromatopsia Management
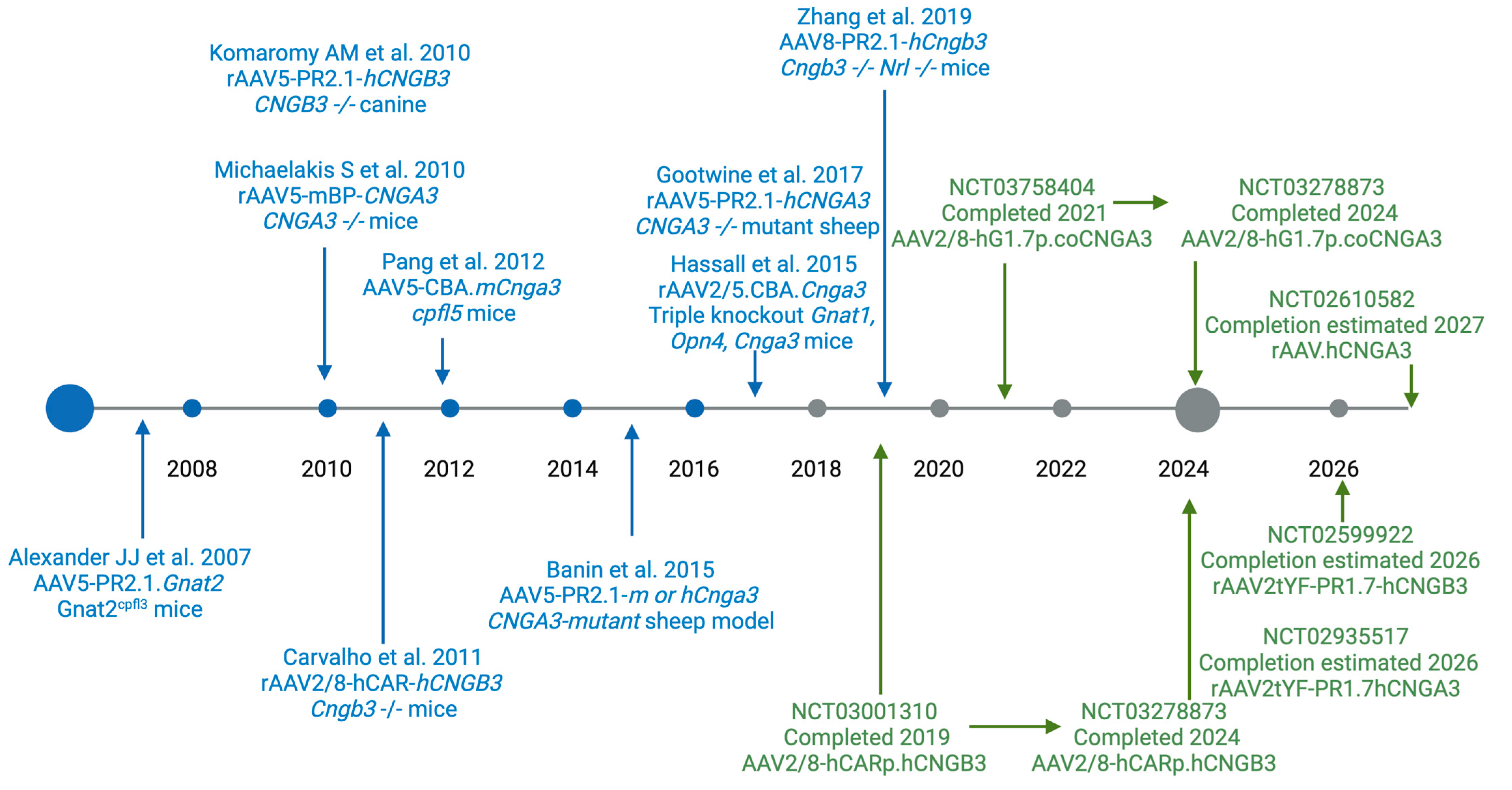
6. Vision Restoration in Pre-Clinical Animal Models of Achromatopsia
7. Gene Therapy Clinical Trials for Achromatopsia
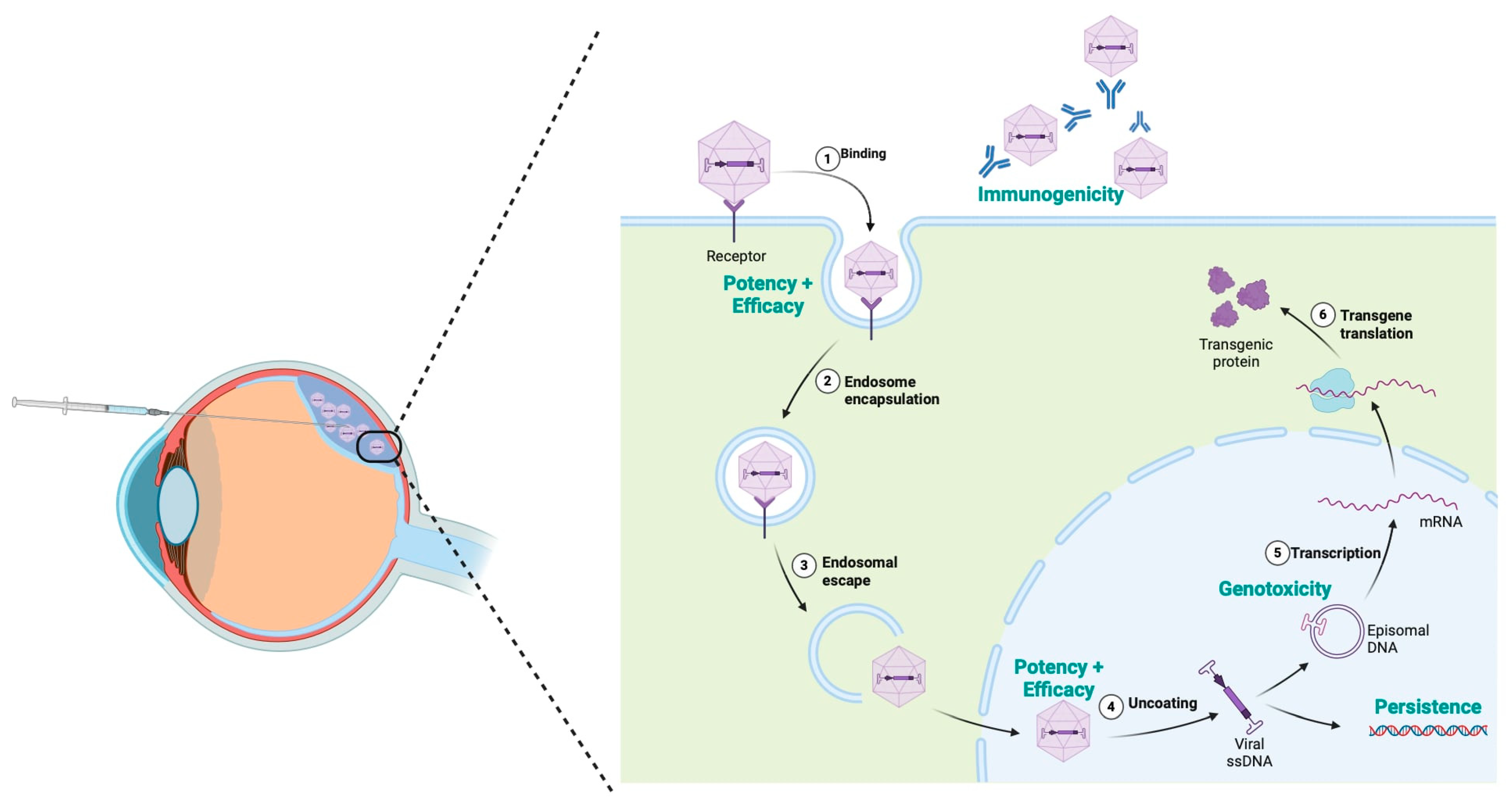
8. Challenges and Limitations
9. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michaelides, M.; Hunt, D.M.; Moore, A.T. The cone dysfunction syndromes. Br. J. Ophthalmol. 2004, 88, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Aboshiha, J.; Dubis, A.M.; Cowing, J.; Fahy, R.T.; Sundaram, V.; Bainbridge, J.W.; Michaelides, M. A prospective longitudinal study of retinal structure and function in achromatopsia. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5733–5743. [Google Scholar] [CrossRef] [PubMed]
- Remmer, M.H.; Rastogi, N.; Ranka, M.P.; Ceisler, E.J. Achromatopsia: A review. Curr. Opin. Ophthalmol. 2015, 26, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Wissinger, B.; Jägle, H.; Kohl, S.; Broghammer, M.; Baumann, B.; Hanna, D.B.; Sharpe, L.T. Human rod monochromacy: Linkage analysis and mapping of a cone photoreceptor expressed candidate gene on chromosome 2q11. Genomics 1998, 51, 325–331. [Google Scholar] [CrossRef]
- Wissinger, B.; Gamer, D.; Jägle, H.; Giorda, R.; Marx, T.; Mayer, S.; Kohl, S. CNGA3 mutations in hereditary cone photoreceptor disorders. Am. J. Hum. Genet. 2001, 69, 722–737. [Google Scholar] [CrossRef]
- Mayer, A.K.; Van Cauwenbergh, C.; Rother, C.; Baumann, B.; Reuter, P.; De Baere, E.; Wissinger, B.; Kohl, S. CNGB3 mutation spectrum including copy number variations in 552 achromatopsia patients. Hum. Mutat. 2017, 38, 1579–1591. [Google Scholar] [CrossRef]
- Johnson, S.; Michaelides, M.; Aligianis, I.A.; Ainsworth, J.R.; Mollon, J.D.; Maher, E.R.; Hunt, D.M. Achromatopsia caused by novel mutations in both CNGA3 and CNGB3. J. Med. Genet. 2004, 41, e20. [Google Scholar] [CrossRef]
- Kohl, S.; Baumann, B.; Rosenberg, T.; Kellner, U.; Lorenz, B.; Vadalà, M.; Wissinger, B. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am. J. Hum. Genet. 2002, 71, 422–425. [Google Scholar] [CrossRef]
- Georgiou, M.; Robson, A.G.; Singh, N.; Pontikos, N.; Kane, T.; Hirji, N.; Michaelides, M. Deep Phenotyping of PDE6C-Associated Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5112–5123. [Google Scholar] [CrossRef]
- Kohl, S.; Coppieters, F.; Meire, F.; Schaich, S.; Roosing, S.; Brennenstuhl, C.; Wissinger, B. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am. J. Hum. Genet. 2012, 91, 527–532. [Google Scholar] [CrossRef]
- Hirji, N.; Aboshiha, J.; Georgiou, M.; Bainbridge, J.; Michaelides, M. Achromatopsia: Clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. 2018, 39, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Galvin, O.; Chi, G.; Brady, L.; Hippert, C.; Del Valle Rubido, M.; Daly, A.; Michaelides, M. The Impact of Inherited Retinal Diseases in the Republic of Ireland (ROI) and the United Kingdom (UK) from a Cost-of-Illness Perspective. Clin. Ophthalmol. 2020, 14, 707–719. [Google Scholar] [CrossRef]
- Hirji, N.; Georgiou, M.; Kalitzeos, A.; Bainbridge, J.W.; Kumaran, N.; Aboshiha, J.; Michaelides, M. Longitudinal Assessment of Retinal Structure in Achromatopsia Patients With Long-Term. Follow-up. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5735–5744. [Google Scholar] [CrossRef]
- Michaelides, M.; Hirji, N.; Wong, S.C.; Besirli, C.G.; Zaman, S.; Kumaran, N.; Bainbridge, J. First-in-Human Gene Therapy Trial of AAV8-hCARp.hCNGB3 in Adults and Children With CNGB3-associated Achromatopsia. Am. J. Ophthalmol. 2023, 253, 243–251. [Google Scholar] [CrossRef]
- Georgiou, M.; Robson, A.G.; Fujinami, K.; de Guimarães, T.A.C.; Fujinami-Yokokawa, Y.; Daich Varela, M.; Michaelides, M. Phenotyping and genotyping inherited retinal diseases: Molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, Leber congenital amaurosis, and cone dysfunction syndromes. Prog. Retin. Eye Res. 2024, 100, 101244. [Google Scholar] [PubMed]
- Sundaram, V.; Wilde, C.; Aboshiha, J.; Cowing, J.; Han, C.; Langlo, C.S.; Michaelides, M. Retinal structure and function in achromatopsia: Implications for gene therapy. Ophthalmology 2014, 121, 234–245. [Google Scholar] [CrossRef]
- Georgiou, M.; Singh, N.; Kane, T.; Robson, A.G.; Kalitzeos, A.; Hirji, N.; Michaelides, M. Photoreceptor Structure in GNAT2-Associated Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2020, 61, 40. [Google Scholar] [CrossRef] [PubMed]
- Langlo, C.S.; Erker, L.R.; Parker, M.; Patterson, E.J.; Higgins, B.P.; Summerfelt, P.; Razeen, M.M.; Collison, F.T.; Fishman, G.A.; Kay, C.N.; et al. Repeatability and longitudinal assessment of foveal cone structure in cngb3-associated achromatopsia. Retina 2017, 37, 1956–1966. [Google Scholar] [CrossRef]
- Dubis, A.M.; Cooper, R.F.; Aboshiha, J.; Langlo, C.S.; Sundaram, V.; Liu, B.; Michaelides, M. Genotype-dependent variability in residual cone structure in achromatopsia: Toward developing metrics for assessing cone health. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7303–7311. [Google Scholar] [CrossRef]
- Mastey, R.R.; Georgiou, M.; Langlo, C.S.; Kalitzeos, A.; Patterson, E.J.; Kane, T.; Carroll, J. Characterization of Retinal Structure in ATF6-Associated Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2631–2640. [Google Scholar] [CrossRef]
- Ansar, M.; Santos-Cortez, R.L.; Saqib, M.A.; Zulfiqar, F.; Lee, K.; Ashraf, N.M.; Leal, S.M. Mutation of ATF6 causes autosomal recessive achromatopsia. Hum. Genet. 2015, 134, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, M.; Litts, K.M.; Kalitzeos, A.; Langlo, C.S.; Kane, T.; Singh, N.; Michaelides, M. Adaptive Optics Retinal Imaging in CNGA3-Associated Achromatopsia: Retinal Characterization, Interocular Symmetry, and Intrafamilial Variability. Investig. Ophthalmol. Vis. Sci. 2019, 60, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Langlo, C.S.; Patterson, E.J.; Higgins, B.P.; Summerfelt, P.; Razeen, M.M.; Erker, L.R.; Parker, M.; Collison, F.T.; Fishman, G.A.; Kay, C.N.; et al. Residual Foveal Cone Structure in CNGB3-Associated Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3984–3995. [Google Scholar] [CrossRef] [PubMed]
- Britten-Jones, A.C.; Thai, L.; Flanagan, J.P.M.; Bedggood, P.A.; Edwards, T.L.; Metha, A.B.; Ayton, L.N. Adaptive optics imaging in inherited retinal diseases: A scoping review of the clinical literature. Surv. Ophthalmol. 2024, 69, 51–66. [Google Scholar] [CrossRef]
- Genead, M.A.; Fishman, G.A.; Rha, J.; Dubis, A.M.; Bonci, D.M.; Dubra, A.; Carroll, J. Photoreceptor structure and function in patients with congenital achromatopsia. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7298–7308. [Google Scholar] [CrossRef]
- Jackson, K.; Vergilio, G.K.; Cooper, R.F.; Ying, G.S.; Morgan, J.I.W. A 2-Year Longitudinal Study of Normal Cone Photoreceptor Density. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1420–1430. [Google Scholar] [CrossRef]
- Michalakis, S.; Gerhardt, M.; Rudolph, G.; Priglinger, S.; Priglinger, C. Achromatopsia: Genetics and Gene Therapy. Mol. Diagn. Ther. 2022, 26, 51–59. [Google Scholar] [CrossRef]
- Mannu, G.S. Retinal phototransduction. Neurosciences 2014, 19, 275–280. [Google Scholar]
- Sun, W.; Li, S.; Xiao, X.; Wang, P.; Zhang, Q. Genotypes and phenotypes of genes associated with achromatopsia: A reference for clinical genetic testing. Mol. Vis. 2020, 26, 588–602. [Google Scholar]
- Burkard, M.; Kohl, S.; Krätzig, T.; Tanimoto, N.; Brennenstuhl, C.; Bausch, A.E.; Ruth, P. Accessory heterozygous mutations in cone photoreceptor CNGA3 exacerbate CNG channel-associated retinopathy. J. Clin. Invest. 2018, 128, 5663–5675. [Google Scholar] [CrossRef]
- Kugler, S.A.; Valmaggia, C.; Sturm, V.; Schorderet, D.F.; Todorova, M.G. Analysis of Suspected Achromatopsia by Multimodal Diagnostic Testing. Klin. Monbl. Augenheilkd. 2023, 240, 1158–1173. [Google Scholar] [CrossRef]
- Ben Simon, G.J.; Abraham, F.A.; Melamed, S. Pingelapese achromatopsia: Correlation between paradoxical pupillary response and clinical features. Br. J. Ophthalmol. 2004, 88, 223–225. [Google Scholar] [CrossRef]
- Winick, J.D.; Blundell, M.L.; Galke, B.L.; Salam, A.A.; Leal, S.M.; Karayiorgou, M. Homozygosity mapping of the Achromatopsia locus in the Pingelapese. Am. J. Hum. Genet. 1999, 64, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Sundin, O.H.; Yang, J.M.; Li, Y.; Zhu, D.; Hurd, J.N.; Mitchell, T.N.; Maumenee, I.H. Genetic basis of total colourblindness among the Pingelapese islanders. Nat. Genet. 2000, 25, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Varsanyi, B.; Antunes, G.A.; Baumann, B.; Hoyng, C.B.; Jägle, H.; Wissinger, B. CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur. J. Hum. Genet. 2005, 13, 302–308. [Google Scholar] [CrossRef]
- Zhang, X.J.; Cote, R.H. Phosphodiesterase 6H, cone-specific inhibitor: Basis Sequence: Mouse. AfCS Nat. Mol. Pages 2011, 2011, A001758. [Google Scholar]
- Cote, R.H.; Gupta, R.; Irwin, M.J.; Wang, X. Photoreceptor Phosphodiesterase (PDE6): Structure, Regulatory Mechanisms, and Implications for Treatment of Retinal Diseases. Adv. Exp. Med. Biol. 2022, 1371, 33–59. [Google Scholar]
- Lee, E.J.; Chiang, W.J.; Kroeger, H.; Bi, C.X.; Chao, D.L.; Skowronska-Krawczyk, D.; Lin, J.H. Multiexon deletion alleles of ATF6 linked to achromatopsia. JCI Insight. 2020, 5, e136041. [Google Scholar] [CrossRef]
- Pascual-Camps, I.; Barranco-Gonzalez, H.; Aviñó-Martínez, J.; Silva, E.; Harto-Castaño, M. Diagnosis and Treatment Options for Achromatopsia: A Review of the Literature. J. Pediatr. Ophthalmol. Strabismus 2018, 55, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Holmström, G.; Bondeson, M.L.; Eriksson, U.; Åkerblom, H.; Larsson, E. ‘Congenital’ nystagmus may hide various ophthalmic diagnoses. Acta Ophthalmol. 2014, 92, 412–416. [Google Scholar] [CrossRef]
- Nasser, F.; Weisschuh, N.; Maffei, P.; Milan, G.; Heller, C.; Zrenner, E.; Kuehlewein, L. Ophthalmic features of cone-rod dystrophy caused by pathogenic variants in the ALMS1 gene. Acta Ophthalmol. 2018, 96, e445–e454. [Google Scholar] [CrossRef] [PubMed]
- Munk, A.H.; Starup, E.B.; Lambon Ralph, M.A.; Leff, A.P.; Starrfelt, R.; Robotham, R.J. Colour perception deficits after posterior stroke: Not so rare after all? Cortex 2023, 159, 118–130. [Google Scholar] [CrossRef]
- Retinal Disorders (Version 4.90): PanelApp. 2024. Available online: https://panelapp.genomicsengland.co.uk/panels/307/ (accessed on 30 August 2024).
- Marian, A.J. Clinical Interpretation and Management of Genetic Variants. JACC Basic. Transl. Sci. 2020, 5, 1029–1042. [Google Scholar] [CrossRef]
- Chen, E.; Facio, F.M.; Aradhya, K.W.; Rojahn, S.; Hatchell, K.E.; Aguilar, S.; Aradhya, S. Rates and Classification of Variants of Uncertain Significance in Hereditary Disease Genetic Testing. JAMA Netw. Open. 2023, 6, e2339571. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.S.; Zouk, H.; Venner, E.; Eng, C.M.; Funke, B.H.; Amendola, L.M.; Carrell, D.S.; Chisholm, R.L.; Chung, W.K.; Denny, J.C.; et al. Frequency of genomic secondary findings among 21,915 eMERGE network participants. Genet. Med. 2020, 22, 1470–1477. [Google Scholar] [CrossRef]
- Christenhusz, G.M.; Devriendt, K.; Dierickx, K. To tell or not to tell? A systematic review of ethical reflections on incidental findings arising in genetics contexts. Eur. J. Hum. Genet. 2013, 21, 248–255. [Google Scholar] [CrossRef]
- ABIM Foundation. Choosing Wisely Campaign. 2023. Available online: https://www.choosingwisely.org/ (accessed on 19 August 2024).
- Aboshiha, J.; Kumaran, N.; Kalitzeos, A.; Hogg, C.; Rubin, G.; Michaelides, M. A Quantitative and Qualitative Exploration of Photoaversion in Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3537–3546. [Google Scholar] [CrossRef]
- Alexander, J.J.; Umino, Y.; Everhart, D.; Chang, B.; Min, S.H.; Li, Q.; Hauswirth, W.W. Restoration of cone vision in a mouse model of achromatopsia. Nat. Med. 2007, 13, 685–687. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Mühlfriedel, R.; Tanimoto, N.; Krishnamoorthy, V.; Koch, S.; Fischer, M.D.; Seeliger, M.W. Restoration of cone vision in the CNGA3-/- mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 2010, 18, 2057–2063. [Google Scholar] [CrossRef]
- Hassall, M.M.; Barnard, A.R.; MacLaren, R.E. Gene Therapy for Color Blindness. Yale J. Biol. Med. 2017, 90, 543–551. [Google Scholar]
- Pang, J.; Wen-Tao, D.; Dai, X.; Lei, B.; Everhart, D.; Umino, Y.; Hauswirth, W.W. AAV-mediated Cone Rescue in a Naturally Occuring Mouse Model of CNGA3-Achromatopsia. PLoS ONE 2012, 7, 4. [Google Scholar] [CrossRef]
- Komáromy, A.M.; Alexander, J.J.; Rowlan, J.S.; Garcia, M.M.; Chiodo, V.A.; Kaya, A.; Aguirre, G.D. Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 2010, 19, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.; Xu, J.; Pearson, R.A.; Smith, A.J.; Bainbridge, J.W.; Morris, L.M.; Ali, R.R. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum. Mol. Genet. 2011, 20, 3161–3175. [Google Scholar] [CrossRef]
- Banin, E.; Gootwine, E.; Obolensky, A.; Ezra-Elia, R.; Ejzenberg, A.; Zelinger, L.; Ofri, R. Gene Augmentation Therapy Restores Retinal Function and Visual Behavior in a Sheep Model of CNGA3 Achromatopsia. Mol. Ther. 2015, 23, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Gootwine, E.; Abu-Siam, M.; Obolensky, A.; Rosov, A.; Honig, H.; Nitzann, T.; Seroussi, E. Gene Augmentation Therapy for a Missense Substitution in the cGMP-Binding Domain of Ovine CNGA3 Gene Restores Vision in Day-Blind Sheep. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1577–1584. [Google Scholar]
- Zhang, Y.; Wang, S.; Xu, M.; Pang, J.; Yuan, Z.; Zhao, C. AAV-mediated human CNGB3 restores cone function in an all-cone mouse model of CNGB3 achromatopsia. J Biomed Res. 2019, 30, 114–121. [Google Scholar] [CrossRef]
- Moshiri, A.; Issa, T.; Rogers, J.; Chen, R.; Thomasy, S.; Stout, T. AAV-mediated gene therapy for PDE6C achromatopsia: Progress and challenges. Investig Ophthalmol Vis Sci. 2024, 65, 4267. [Google Scholar]
- Reichel, F.F.; Michalakis, S.; Wilhelm, B.; Zobor, D.; Muehlfriedel, R.; Kohl, S.; Fischer, D. Three-year results of phase I retinal gene therapy trial for CNGA3-mutated achromatopsia: Results of a non randomised controlled trial. Br. J. Ophthalmol. 2022, 106, 1567–1572. [Google Scholar] [CrossRef]
- Fischer, M.D.; Michalakis, S.; Wilhelm, B.; Zobor, D.; Muehlfriedel, R.; Kohl, S.; Wissinger, B. Safety and Vision Outcomes of Subretinal Gene Therapy Targeting Cone Photoreceptors in Achromatopsia: A Nonrandomized Controlled Trial. JAMA Ophthalmol. 2020, 138, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Zhu, X.; Hong, J.; Zhou, X. Biological Immune Mechanism of Retina. Front. Biosci. 2023, 28, 363. [Google Scholar] [CrossRef]
- Drag, S.; Dotiwala, F.; Upadhyay, A.K. Gene Therapy for Retinal Degenerative Diseases: Progress, Challenges, and Future Directions. Investig. Ophthalmol. Vis. Sci. 2023, 64, 39. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, N.; Parachuri, N.; Bandello, F.; Kuppermann, B.D. Immunogenicity: Clouding the Future of Intravitreal Therapy. Ocul. Immunol. Inflamm. 2023, 31, 1746–1749. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Li, D.; Wang, N.; Gruber, J.; Lo, A.W.; Conti, R.M. The estimated annual financial impact of gene therapy in the United States. Gene Ther. 2023, 30, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, M. Should rare diseases get special treatment? J. Med. Ethics. 2022, 48, 86–92. [Google Scholar] [CrossRef]
- Samiy, N. Gene therapy for retinal diseases. J. Ophthalmic Vis. Res. 2014, 9, 506–509. [Google Scholar] [CrossRef]
- Molz, B.; Herbik, A.; Baseler, H.A.; de Best, P.; Raz, N.; Gouws, A.; Hoffmann, M.B. Achromatopsia—Visual Cortex Stability and Plasticity in the Absence of Functional Cones. Investig. Ophthalmol. Vis. Sci. 2023, 64, 23. [Google Scholar] [CrossRef]
- Peddi, N.C.; Marasandra Ramesh, H.; Gude, S.S.; Gude, S.S.; Vuppalapati, S. Intrauterine Fetal Gene Therapy: Is That the Future and Is That Future Now? Cureus 2022, 14, e22521. [Google Scholar] [CrossRef]
- Mühlfriedel, R.; Tanimoto, N.; Schön, C.; Sothilingam, V.; Garcia Garrido, M.; Beck, S.C.; Michalakis, S. AAV-Mediated Gene Supplementation Therapy in Achromatopsia Type 2: Preclinical Data on Therapeutic Time Window and Long-Term Effects. Front. Neurosci. 2017, 11, 292. [Google Scholar] [CrossRef]
- Zein, W.M.; Jeffrey, B.G.; Wiley, H.E.; Turriff, A.E.; Tumminia, S.J.; Tao, W.; Sieving, P.A. CNGB3-achromatopsia clinical trial with CNTF: Diminished rod pathway responses with no evidence of improvement in cone function. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6301–6308. [Google Scholar] [CrossRef]
- Fischer, M.D.; Simonelli, F.; Sahni, J.; Holz, F.G.; Maier, R.; Fasser, C.; Leroy, B.P. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules 2024, 14, 122. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Gene | CNGA3 | CNGB3 | GNAT2 | PDE6C | PDE6H | ATF6 |
|---|---|---|---|---|---|---|
| Location | 2q11.2 | 8q21.3 | 1p13.3 | 10q23.33 | 12p12.3 | 1q23.3 |
| CDS length (base pairs) | 2085 | 2430 | 1065 | 2577 | 252 | 2013 |
| Protein encoded | Encodes alpha-subunit of cyclic nucleotide-gated (CNG) channel | Encodes beta-subunit of cyclic nucleotide-gated (CNG) channel | Encodes G protein alpha-subunit of transducin 2 | Encodes alpha catalytic subunit of cone photoreceptor phosphodiesterase | Encodes gamma catalytic subunit of cone photoreceptor phosphodiesterase | Encodes for activating transcription factor 6 |
| Fundus autofluorescence | Normal appearance, central increased signal, central decreased signal [2] | Normal appearance, central increased signal, central decreased signal [2] | Typically, normal appearance [17] | Fundus autofluorescence shows decreased central signal with a surrounding hyperautofluorescence [9] | Normal fundus autofluorescence [10] | Variable changes; increased or decreased autofluorescence ring [8] |
| OCT | Foveal hypoplasia in 60–70% of achromatopsia [2]; OCT 50% grade I to III | Foveal hypoplasia in 60–70% of achromatopsia [2]; OCT 50% grade I to III [18] | Foveal hypoplasia was not seen; [19] OCT typically grade I [17] | No foveal hypoplasia; no OCT of grade I and II [9] | No foveal hypoplasia; preserved OCT [10] | Foveal hypoplasia is present in all patients; no reported grades I and II [20] |
| Electrophysiology | ffERG severely reduced cone response with normal/subnormal rod response | ffERG severely reduced cone response with normal/subnormal rod response | ffERG severely reduced cone response; relatively preserved S-cone compared to CNGA3 and CNGB3 [17] | Some degree of preserved S-cone similar to GNAT2; scotopic ERG can show mild-moderate decrease [17] | Some degree of preserved S-cone similar to GNAT2 [15] | ffERG severely reduced amplitude of cone response [21] |
| AOSLO | Marked variability [22] | Marked variability [23] | Least disrupted photoreceptor mosaic and reflectivity preserved [17] | Few, if any, cellular residual structures [9] | Not well characterised | Few, if any, remnant cone structures [20] |
| NCT Registration | Phase | Gene | Capsid | Vector | Sponsor | Status | Route |
|---|---|---|---|---|---|---|---|
| 03001310 03278873 | I/II | CNGB3 | AAV5 | AAV2/8-hCARp.hCNGB3 | MeiraGTx/ Janssen | Completed; active | Subretinal |
| 03758404 03278873 | I/II | CNGA3 | AAV5 | AAV2/8-hG1.7p.coCNGA3 | MeiraGTx/ Janssen | Completed; active | Subretinal |
| 02610582 | I/II | CNGA3 | rAAV8 | rAAV.hCNGA3 | RD-CURE | Recruiting | Subretinal |
| 02599922 | I/II | CNGB3 | AAV2tYF | rAAV2tYF-PR1.7-hCNGB3 (AGTC-401) | AGTC | Active | Subretinal |
| 02935517 | I/II | CNGA3 | AAV2tYF | rAAV2tYF-PR1.7hCNGA3 (AGTC-402) | AGTC | Active | Subretinal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baxter, M.F.; Borchert, G.A. Gene Therapy for Achromatopsia. Int. J. Mol. Sci. 2024, 25, 9739. https://doi.org/10.3390/ijms25179739
Baxter MF, Borchert GA. Gene Therapy for Achromatopsia. International Journal of Molecular Sciences. 2024; 25(17):9739. https://doi.org/10.3390/ijms25179739
Chicago/Turabian StyleBaxter, Megan F., and Grace A. Borchert. 2024. "Gene Therapy for Achromatopsia" International Journal of Molecular Sciences 25, no. 17: 9739. https://doi.org/10.3390/ijms25179739