
GWAS Enhances Genomic Prediction Accuracy of Caviar Yield, Caviar Color and Body Weight Traits in Sturgeons Using Whole-Genome Sequencing Data

 ,
,
Abstract
:1. Introduction
2. Results
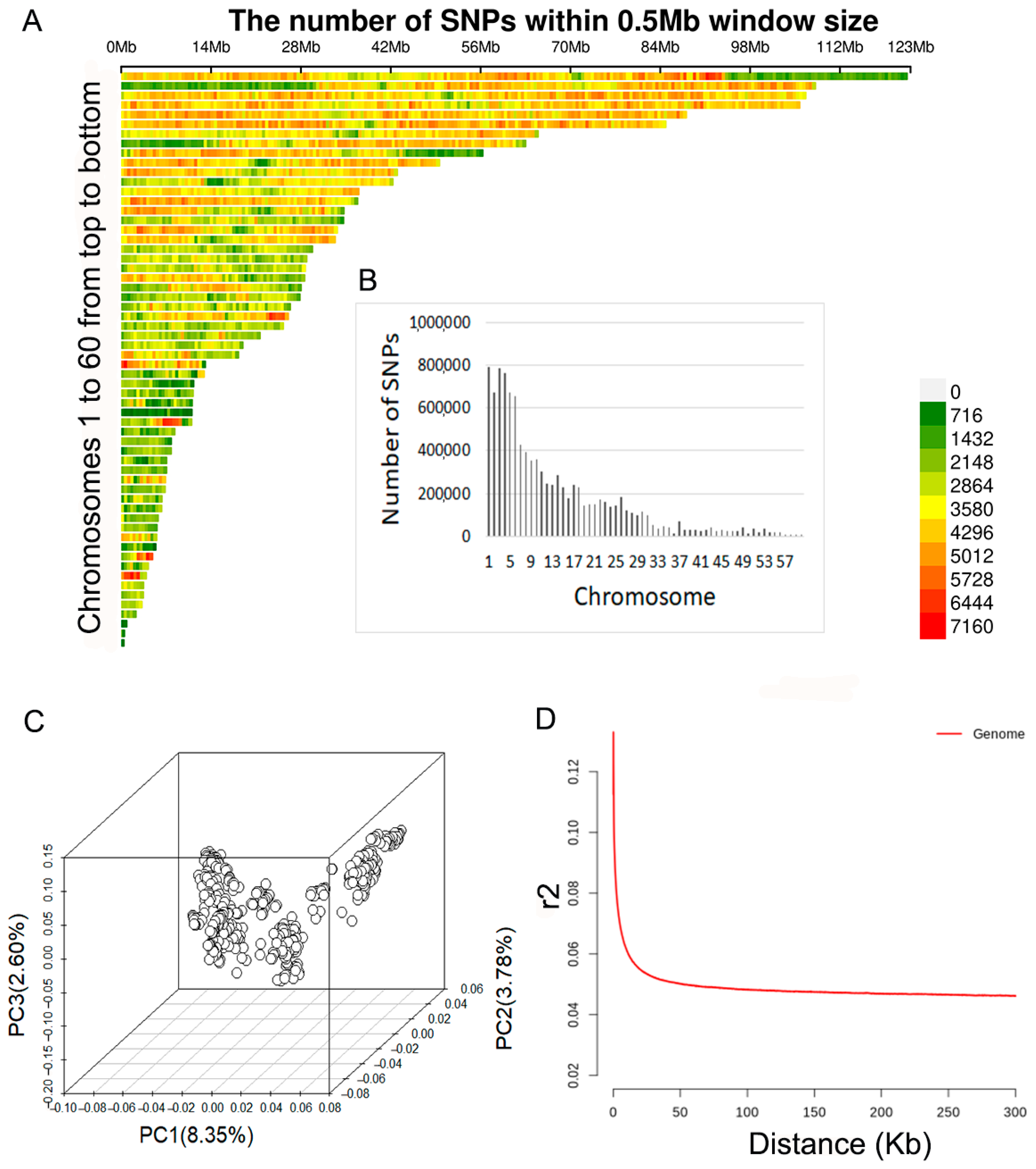
2.1. Whole-Genome Sequencing and SNP Calling
2.2. Population Structure Analysis
2.3. Phenotype Statistics and Heritability Estimation
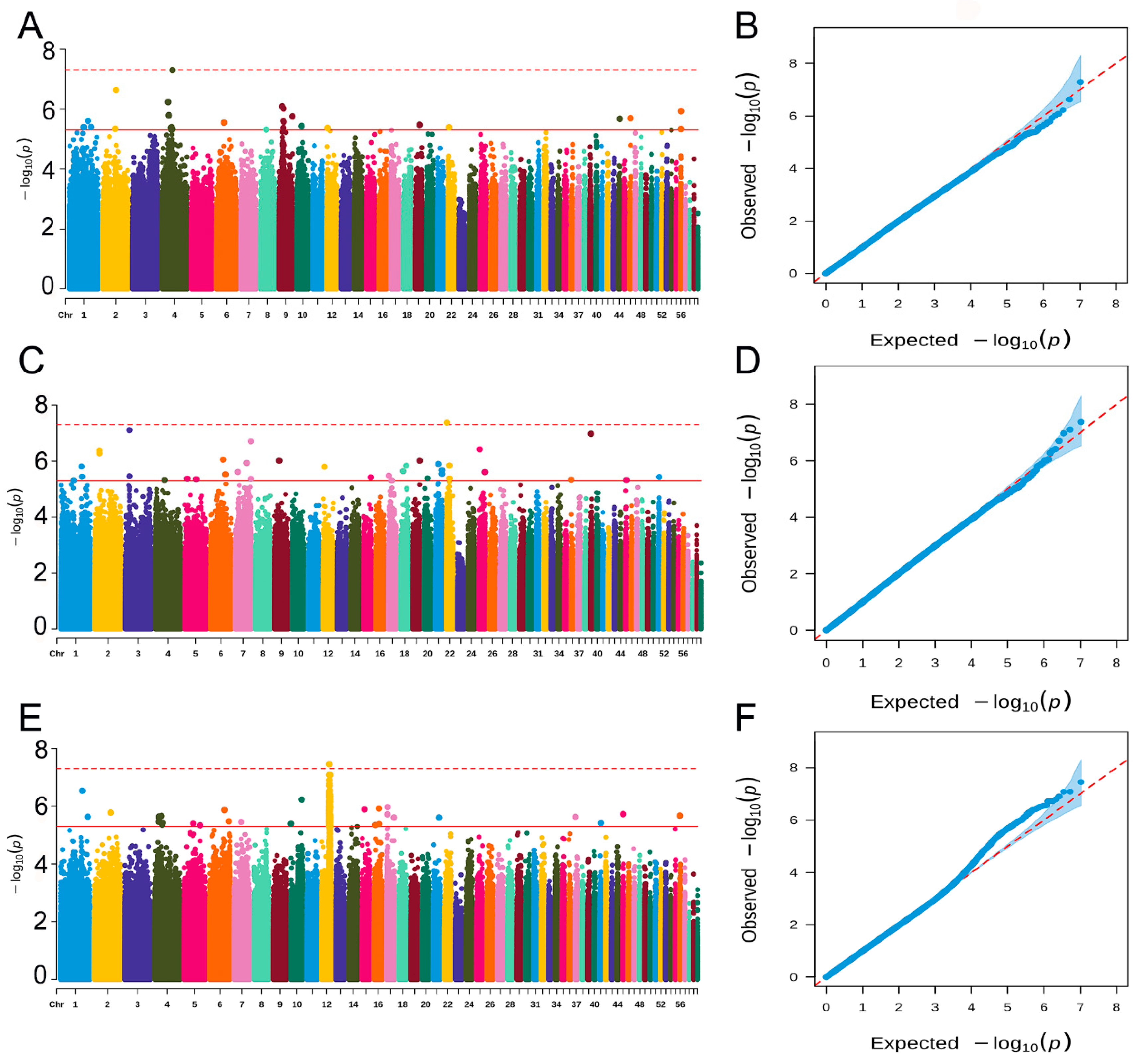
2.4. Genome-Wide Association Study
2.5. Identification of Candidate Genes
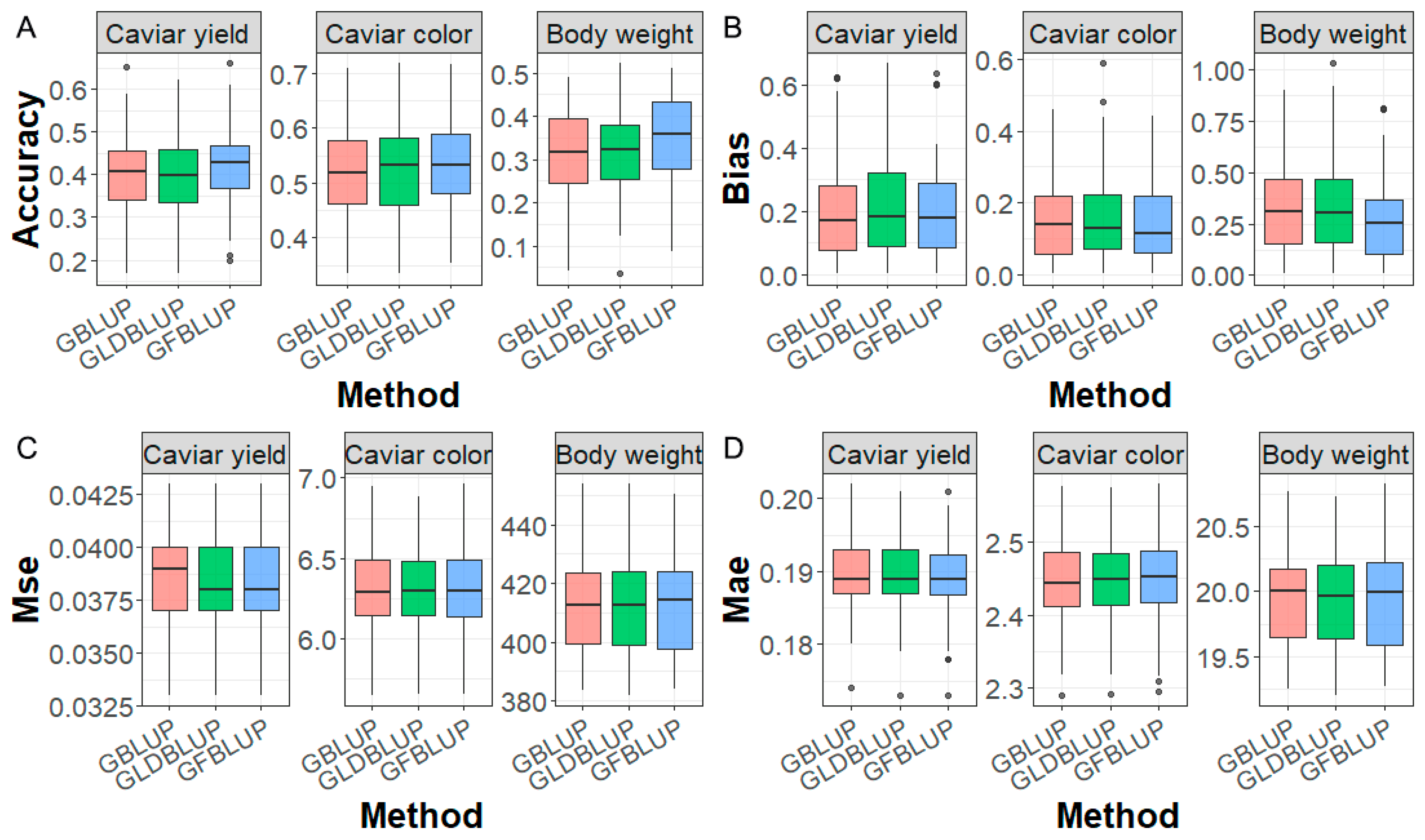
2.6. Genomic Prediction Performance
3. Discussion
3.1. Potential Candidate Genes for Caviar Yield
3.2. Potential Candidate Genes for Caviar Color
3.3. Potential Candidate Genes for Body Weight
3.4. Genomic Prediction Incorporating GWAS Prior Information
4. Materials and Methods
4.1. Population and Phenotyping Measurement
4.2. Whole-Genome Sequencing
4.3. Genotype Imputation and Population Structure Analysis
4.4. Genome-Wide Association Study
4.5. Functional Genomic Analysis
4.6. Genomic Prediction Incorporating GWAS Prior Information
4.6.1. GBLUP
4.6.2. GFBLUP
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| SNPs | Single nucleotide polymorphisms |
| GWAS | Genome-wide association study |
| GFBLUP | Genomic feature BLUP |
| LD | Linkage disequilibrium |
| CITES | Convention on International Trade in Endangered Species of Wild Fauna and Flora |
| GS | Genomic selection |
| GEBV | Genomic estimated breeding values |
| QTLs | Quantitative trait locis |
| PIT | Passive integrated transponder |
| BW | Body weight |
| CW | Caviar weight |
| CC | Caviar color |
| CY | Caviar yield |
| DP | Depth |
| QUAL | Quality |
| QD | Quality by depth |
| MAF | Minor allele frequency |
| HWE | Hardy-Weinberg equilibrium |
| PCA | Principal component analysis |
| PCs | Principal components |
| Quantile-quantile | |
| GBLUP | Genomic best linear unbiased prediction |
| CV | Cross-validation |
| Mse | Mean squared error |
| Mae | Mean absolute error |
References
- Du, K.; Stock, M.; Kneitz, S.; Klopp, C.; Woltering, J.M.; Adolfi, M.C.; Feron, R.; Prokopov, D.; Makunin, A.; Kichigin, I.; et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 2020, 4, 841–852. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lee, J.S.; Dang, C.G.; Sudrajad, P.; Kim, H.C.; Yeon, S.H.; Kang, H.S.; Lee, S.H. Stories and Challenges of Genome Wide Association Studies in Livestock—A Review. Asian-Australas. J. Anim. Sci. 2015, 28, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef]
- Yáñez, J.M.; Barría, A.; López, M.E.; Moen, T.; Garcia, B.F.; Yoshida, G.M.; Xu, P. Genome-wide association and genomic selection in aquaculture. Rev. Aquacult. 2023, 15, 645–675. [Google Scholar] [CrossRef]
- Zheng, X.H.; Kuang, Y.Y.; Lv, W.H.; Cao, D.C.; Sun, Z.P.; Sun, X.W. Genome-Wide Association Study for Muscle Fat Content and Abdominal Fat Traits in Common Carp (Cyprinus carpio). PLoS ONE 2016, 11, e0169127. [Google Scholar] [CrossRef]
- Horn, S.S.; Ruyter, B.; Meuwissen, T.H.E.; Moghadam, H.; Hillestad, B.; Sonesson, A.K. GWAS identifies genetic variants associated with omega-3 fatty acid composition of Atlantic salmon fillets. Aquaculture 2020, 514, 734494. [Google Scholar] [CrossRef]
- Xiao, S.J.; Wang, P.P.; Dong, L.S.; Zhang, Y.G.; Han, Z.F.; Wang, Q.R.; Wang, Z.Y. Whole-genome single-nucleotide polymorphism (SNP) marker discovery and association analysis with the eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) content in Larimichthys crocea. PeerJ 2016, 4, e2664. [Google Scholar] [CrossRef]
- Li, N.; Zhou, T.; Geng, X.; Jin, Y.L.; Wang, X.Z.; Liu, S.K.; Xu, X.Y.; Gao, D.Y.; Li, Q.; Liu, Z.J. Identification of novel genes significantly affecting growth in catfish through GWAS analysis. Mol. Genet. Genom. 2018, 293, 587–599. [Google Scholar] [CrossRef]
- Geng, X.; Liu, S.K.; Yao, J.; Bao, L.S.; Zhang, J.R.; Li, C.; Wang, R.J.; Sha, J.; Zeng, P.; Zhi, D.G.; et al. A Genome-Wide Association Study Identifies Multiple Regions Associated with Head Size in Catfish. G3 Genes Genomes Genet. 2016, 6, 3389–3398. [Google Scholar] [CrossRef]
- Robledo, D.; Matika, O.; Hamilton, A.; Houston, R.D. Genome-Wide Association and Genomic Selection for Resistance to Amoebic Gill Disease in Atlantic Salmon. G3 Genes Genomes Genet. 2018, 8, 1195–1203. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, Z.; Pan, Y.; Zhao, J.; Bai, H.; Chen, B.; Zhang, X.; Pu, F.; Chen, J.; Xu, P. GWAS identified candidate variants and genes associated with acute heat tolerance of large yellow croaker. Aquaculture 2021, 540, 736696. [Google Scholar] [CrossRef]
- Zeng, J.; Zhao, J.; Wang, J.; Bai, Y.; Long, F.; Deng, Y.; Jiang, P.; Xiao, J.; Qu, A.; Tong, B.; et al. Genetic linkage between swimming performance and disease resistance enables multitrait breeding strategies in large yellow croaker. Agric. Commun. 2023, 1, 100019. [Google Scholar] [CrossRef]
- Yoshida, G.M.; Yáñez, J.M. Increased accuracy of genomic predictions for growth under chronic thermal stress in rainbow trout by prioritizing variants from GWAS using imputed sequence data. Evol. Appl. 2021, 15, 537–552. [Google Scholar] [CrossRef]
- Song, H.; Li, L.; Ma, P.; Zhang, S.; Su, G.; Lund, M.S.; Zhang, Q.; Ding, X. Short communication: Improving the accuracy of genomic prediction of body conformation traits in Chinese Holsteins using markers derived from high-density marker panels. J. Dairy Sci. 2018, 101, 5250–5254. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Zhou, F.; Yao, Z.; Zhan, Y.; Fan, Z.; Meng, X.; Zhang, Z.; Liu, L.; Yang, J.; et al. Increased Accuracy of Genomic Prediction Using Preselected SNPs from GWAS with Imputed Whole-Genome Sequence Data in Pigs. Animals 2023, 13, 3871. [Google Scholar] [CrossRef] [PubMed]
- Song, H.L.; Ye, S.P.; Jiang, Y.F.; Zhang, Z.; Zhang, Q.; Ding, X.D. Using imputation-based whole-genome sequencing data to improve the accuracy of genomic prediction for combined populations in pigs. Genet. Sel. Evol. 2019, 51, 58. [Google Scholar] [CrossRef]
- Song, H.; Hu, H. Strategies to improve the accuracy and reduce costs of genomic prediction in aquaculture species. Evol. Appl. 2021, 15, 578–590. [Google Scholar] [CrossRef]
- Lu, S.; Liu, Y.; Yu, X.J.; Li, Y.Z.; Yang, Y.M.; Wei, M.; Zhou, Q.; Wang, J.; Zhang, Y.P.; Zheng, W.W.; et al. Prediction of genomic breeding values based on pre-selected SNPs using ssGBLUP, WssGBLUP and BayesB for Edwardsiellosis resistance in Japanese flounder. Genet. Sel. Evol. 2020, 52, 49. [Google Scholar] [CrossRef]
- Lin, J.; Ji, Z.; Di, Z.; Zhang, Y.; Yan, C.; Zeng, S. Overexpression of Tfap2a in Mouse Oocytes Impaired Spindle and Chromosome Organization. Int. J. Mol. Sci. 2022, 23, 14376. [Google Scholar] [CrossRef]
- Niu, X.; Huang, Y.; Lu, H.; Li, S.; Huang, S.; Ran, X.; Wang, J. CircRNAs in Xiang pig ovaries among diestrus and estrus stages. Porc. Health Manag. 2022, 8, 29. [Google Scholar] [CrossRef]
- Yin, Y.; Sheng, J.; Hu, R.; Yang, Y.; Qing, S. The expression and localization of Crb3 in developmental stages of the mice embryos and in different organs of 1-week-old female mice. Reprod. Domest. Anim. 2014, 49, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, L.; Zhou, H.-X.; Liu, L.; Lu, A.; Li, G.-P.; Schatten, H.; Liang, C.-G. Clathrin Heavy Chain 1 is Required for Spindle Assembly and Chromosome Congression in Mouse Oocytes. Microsc. Microanal. 2013, 19, 1364–1373. [Google Scholar] [CrossRef]
- Cloutier, J.M.; Mahadevaiah, S.K.; ElInati, E.; Nussenzweig, A.; Toth, A.; Turner, J.M. Histone H2AFX Links Meiotic Chromosome Asynapsis to Prophase I Oocyte Loss in Mammals. PLoS Genet. 2015, 11, e1005462. [Google Scholar] [CrossRef]
- Liu, J.; Qi, N.; Xing, W.; Li, M.; Qian, Y.; Luo, G.; Yu, S. The TGF-beta/SMAD Signaling Pathway Prevents Follicular Atresia by Upregulating MORC2. Int. J. Mol. Sci. 2022, 23, 10657. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodi, M.; Cheraghi, E.; Riahi, A. The Effect of Wharton’s Jelly-Derived Conditioned Medium on the In Vitro Maturation of Immature Oocytes, Embryo Development, and Genes Expression Involved in Apoptosis. Reprod. Sci. 2024, 31, 190–198. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, S.Y.; Lee, H.S.; Kim, E.Y.; Ko, J.J.; Lee, K.A. Zap70 and downstream RanBP2 are required for the exact timing of the meiotic cell cycle in oocytes. Cell Cycle 2017, 16, 1534–1546. [Google Scholar] [CrossRef]
- Shen, Y.; Ulaangerel, T.; Ren, H.; Davshilt, T.; Yi, M.; Li, X.; Xing, J.; Du, M.; Bai, D.; Dugarjav, M.; et al. Proteomic Differences Between the Ovulatory and Anovulatory Sides of the Mare’s Follicular and Oviduct Fluid. J. Equine Vet. Sci. 2023, 121, 104207. [Google Scholar] [CrossRef]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Co-expression analysis and identification of fecundity-related long non-coding RNAs in sheep ovaries. Sci. Rep. 2016, 6, 39398. [Google Scholar]
- Cha, J.; Jin, D.; Kim, J.H.; Kim, S.C.; Lim, J.A.; Chai, H.H.; Jung, S.A.; Lee, J.H.; Lee, S.H. Genome-wide association study revealed the genomic regions associated with skin pigmentation in an Ogye x White Leghorn F2 chicken population. Poult. Sci. 2023, 102, 102720. [Google Scholar] [CrossRef]
- Batai, K.; Cui, Z.; Arora, A.; Shah-Williams, E.; Hernandez, W.; Ruden, M.; Hollowell, C.M.P.; Hooker, S.E.; Bathina, M.; Murphy, A.B.; et al. Genetic loci associated with skin pigmentation in African Americans and their effects on vitamin D deficiency. PLoS Genet. 2021, 17, e1009319. [Google Scholar] [CrossRef] [PubMed]
- Mussig, C.; Schroder, F.; Usadel, B.; Lisso, J. Structure and putative function of NFX1-like proteins in plants. Plant Biol. 2010, 12, 381–394. [Google Scholar] [CrossRef]
- Fiil, B.K.; Damgaard, R.B.; Wagner, S.A.; Keusekotten, K.; Fritsch, M.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C.; Komander, D.; Gyrd-Hansen, M. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol. Cell. 2013, 50, 818–830. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, J. Potential molecular characteristics in situ in response to repetitive UVB irradiation. Diagn. Pathol. 2016, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- April, C.S.; Barsh, G.S. Distinct pigmentary and melanocortin 1 receptor-dependent components of cutaneous defense against ultraviolet radiation. PLoS Genet. 2007, 3, e9. [Google Scholar] [CrossRef]
- Linher-Melville, K.; Li, J.L. The roles of glial cell line-derived neurotrophic factor, brain-derived neurotrophic factor and nerve growth factor during the final stage of folliculogenesis: A focus on oocyte maturation. Reproduction 2013, 145, R43–R54. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Mo, D.; Li, A.; Gong, W.; Xiao, S.; Zhang, Y.; Qin, L.; Niu, Y.; Guo, Y.; Liu, X.; et al. Comparative Analyses by Sequencing of Transcriptomes during Skeletal Muscle Development between Pig Breeds Differing in Muscle Growth Rate and Fatness. PLoS ONE 2011, 6, e19774. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, X.; Wang, W.; Zhang, D.; Li, X.; Zhang, Y.; Zhao, Y.; Cheng, J.; Zhao, L.; Wang, J.; et al. Detection of single nucleotide polymorphism in HTR4 and its relationship with growth traits in sheep. Anim. Biotechnol. 2023, 34, 4600–4607. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, C.; Han, S.; Chen, L.; Ding, H.; Lin, Y.; Zhang, G.; Xie, K.; Wang, J.; Dai, G. Integrated Analysis Reveals a lncRNA-miRNA-mRNA Network Associated with Pigeon Skeletal Muscle Development. Genes 2021, 12, 1787. [Google Scholar] [CrossRef]
- Orkunoglu-Suer, F.E.; Gordish-Dressman, H.; Clarkson, P.M.; Thompson, P.D.; Angelopoulos, T.J.; Gordon, P.M.; Moyna, N.M.; Pescatello, L.S.; Visich, P.S.; Zoeller, R.F.; et al. INSIG2 gene polymorphism is associated with increased subcutaneous fat in women and poor response to resistance training in men. BMC Med. Genet. 2008, 9, 117. [Google Scholar] [CrossRef]
- Bao, G.; Li, S.; Zhao, F.; Wang, J.; Liu, X.; Hu, J.; Shi, B.; Wen, Y.; Zhao, L.; Luo, Y. Comprehensive Transcriptome Analysis Reveals the Role of lncRNA in Fatty Acid Metabolism in the Longissimus Thoracis Muscle of Tibetan Sheep at Different Ages. Front. Nutr. 2022, 9, 847077. [Google Scholar] [CrossRef]
- Song, Y.; Ahn, J.; Suh, Y.; Davis, M.E.; Lee, K. Identification of novel tissue-specific genes by analysis of microarray databases: A human and mouse model. PLoS ONE 2013, 8, e64483. [Google Scholar] [CrossRef] [PubMed]
- Avirneni-Vadlamudi, U.; Galindo, K.A.; Endicott, T.R.; Paulson, V.; Cameron, S.; Galindo, R.L. Drosophila and mammalian models uncover a role for the myoblast fusion gene TANC1 in rhabdomyosarcoma. J. Clin. Investig. 2012, 122, 403–407. [Google Scholar] [CrossRef]
- Xie, Q.; Zhang, Z.; Chen, Z.; Sun, J.; Li, M.; Wang, Q.; Pan, Y. Integration of Selection Signatures and Protein Interactions Reveals NR6A1, PAPPA2, and PIK3C2B as the Promising Candidate Genes Underlying the Characteristics of Licha Black Pig. Biology 2023, 12, 500. [Google Scholar] [CrossRef]
- Schumann, T.; Konig, J.; von Loeffelholz, C.; Vatner, D.F.; Zhang, D.; Perry, R.J.; Bernier, M.; Chami, J.; Henke, C.; Kurzbach, A.; et al. Deletion of the diabetes candidate gene Slc16a13 in mice attenuates diet-induced ectopic lipid accumulation and insulin resistance. Commun. Biol. 2021, 4, 826. [Google Scholar]
- Terakado, A.P.N.; Costa, R.B.; de Camargo, G.M.F.; Irano, N.; Bresolin, T.; Takada, L.; Carvalho, C.V.D.; Oliveira, H.N.; Carvalheiro, R.; Baldi, F.; et al. Genome-wide association study for growth traits in Nelore cattle. Animal 2018, 12, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Ramayo-Caldas, Y.; Ballester, M.; Fortes, M.R.S.; Esteve-Codina, A.; Castelló, A.; Noguera, J.L.; Fernández, A.I.; Pérez-Enciso, M.; Reverter, A.; Folch, J.M. From SNP co-association to RNA co-expression: Novel insights into gene networks for intramuscular fatty acid composition in porcine. BMC Genom. 2014, 15, 232. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Zhang, L.; Yu, M.; Sheng, Z.; Shi, M.; Zhu, Y.; Zhang, Z.; Bo, P. Activiated galanin receptor 2 attenuates insulin resistance in skeletal muscle of obese mice. Peptides 2018, 99, 92–98. [Google Scholar] [CrossRef]
- Mohammadabadi, M.; Bordbar, F.; Jensen, J.; Du, M.; Guo, W. Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals. Animals 2021, 11, 835. [Google Scholar] [CrossRef]
- Goh, B.C.; Singhal, V.; Herrera, A.J.; Tomlinson, R.E.; Kim, S.; Faugere, M.C.; Germain-Lee, E.L.; Clemens, T.L.; Lee, S.J.; DiGirolamo, D.J. Activin receptor type 2A (ACVR2A) functions directly in osteoblasts as a negative regulator of bone mass. J. Biol. Chem. 2017, 292, 13809–13822. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, J.; Li, Y.; Ahmad, H.I.; Li, T.; Liang, Q.; Li, Y.; Yang, M.; Han, J. Liver Transcriptome Profiling Identifies Key Genes Related to Lipid Metabolism in Yili Geese. Animals 2023, 13, 3473. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, X.; Li, F.; La, Y.; Li, G.; Zhang, Y.; Li, X.; Zhao, Y.; Song, Q.; Wang, W. The association of polymorphisms in the ovine PPARGC1B and ZEB2 genes with body weight in Hu sheep. Anim. Biotechnol. 2022, 33, 90–97. [Google Scholar] [CrossRef]
- Iheshiulor, O.O.M.; Woolliams, J.A.; Yu, X.J.; Wellmann, R.; Meuwissen, T.H.E. Within- and across-breed genomic prediction using whole-genome sequence and single nucleotide polymorphism panels. Genet. Sel. Evol. 2016, 48, 15. [Google Scholar] [CrossRef]
- Zhu, D.; Zhao, Y.; Zhang, R.; Wu, H.; Cai, G.; Wu, Z.; Wang, Y.; Hu, X. Genomic prediction based on selective linkage disequilibrium pruning of low-coverage whole-genome sequence variants in a pure Duroc population. Genet. Sel. Evol. 2023, 55, 72. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Gao, N.; Zheng, R.; Chen, Z.; Teng, J.; Yuan, X.; Zhang, H.; Chen, Z.; Zhang, X.; Li, J.; et al. Strategies for Obtaining and Pruning Imputed Whole-Genome Sequence Data for Genomic Prediction. Front. Genet. 2019, 10, 673. [Google Scholar] [CrossRef]
- Song, H.; Dong, T.; Wang, W.; Jiang, B.; Yan, X.; Geng, C.; Bai, S.; Xu, S.; Hu, H. Cost-effective genomic prediction of critical economic traits in sturgeons through low-coverage sequencing. Genomics 2024, 116, 110874. [Google Scholar] [CrossRef]
- Song, H.; Xu, S.; Luo, K.; Hu, M.; Luan, S.; Shao, H.; Kong, J.; Hu, H. Estimation of genetic parameters for growth and egg related traits in Russian sturgeon (Acipenser gueldenstaedtii). Aquaculture 2022, 546, 737299. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Browning, B.L.; Browning, S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 2009, 84, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Pe’er, I.; Yelensky, R.; Altshuler, D.; Daly, M.J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. 2008, 32, 381–385. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Shi, L.; Zhang, P.; Li, Y.; Li, M.; Tian, J.; Wang, L.; Zhao, F. GWAS of Reproductive Traits in Large White Pigs on Chip and Imputed Whole-Genome Sequencing Data. Int. J. Mol. Sci. 2022, 23, 13338. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Edwards, S.M.; Sorensen, I.F.; Sarup, P.; Mackay, T.F.C.; Sorensen, P. Genomic Prediction for Quantitative Traits Is Improved by Mapping Variants to Gene Ontology Categories in Drosophila melanogaster. Genetics 2016, 203, 1871–1873. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Trait | Number | Mean | SD | CV | Max | Min |
|---|---|---|---|---|---|---|
| Caviar yield | 673 | 0.190 | 0.057 | 30.00% | 0.439 | 0.021 |
| Caviar color | 673 | 2.453 | 0.653 | 26.62% | 4 | 1 |
| Body weight | 673 | 19.933 | 4.029 | 20.21% | 35.400 | 10.400 |
| Trait | V(G) | V(e) | h2 |
|---|---|---|---|
| Caviar yield | 0.00159 | 0.00161 | 0.497 |
| Caviar color | 0.242 | 0.152 | 0.614 |
| Body weight | 11.493 | 6.844 | 0.627 |
| Chr | SNP_R (bp) | SNP_N | Position_Top (bp) | p Value_Top | Candidate Gene |
|---|---|---|---|---|---|
| 4 | 43,712,625–43,752,625 | 2 | 43,733,654 | 5.12 × 10⁻8 | TFAP2A |
| 4 | 25,562,496–25,602,496 | 1 | 25,582,496 | 5.87 × 10⁻7 | C8orf34 |
| 56 | 2,391,026–2,431,026 | 1 | 2,411,026 | 1.18 × 10⁻6 | PCOLCE, PFN2, RNF167 |
| 9 | 55,004,692–55,044,692 | 1 | 55,024,692 | 1.76 × 10⁻6 | RPS6KA3 |
| 46 | 4,382,490–4,422,490 | 1 | 4,402,490 | 2.03 × 10⁻6 | CRB3, DENND1C, TUBB |
| 44 | 5,849,293–5,889,293 | 1 | 5,869,293 | 2.12 × 10⁻6 | ARCN1, H2AFX, HMBS |
| 1 | 78,013,802–78,053,802 | 1 | 78,033,802 | 2.49 × 10⁻6 | RORB |
| 9 | 17,568,460–17,608,460 | 1 | 17,588,460 | 2.49 × 10⁻6 | SLC5A7, STK24 |
| 9 | 19,246,701–19,286,701 | 1 | 19,266,701 | 2.67 × 10⁻6 | SETD4, morc3 |
| 4 | 40,684,761–40,724,761 | 1 | 40,704,761 | 3.98 × 10⁻6 | BAG1, C7orf25 |
| 22 | 3,969,669–4,009,669 | 1 | 3,989,669 | 4.10 × 10⁻6 | DPY19L3, ZNF507 |
| 4 | 36,848,652–36,888,652 | 1 | 36,868,652 | 4.20 × 10⁻6 | ABHD3 |
| 9 | 17,393,602–17,433,602 | 1 | 17,413,602 | 4.25 × 10⁻6 | RANBP2 |
| 12 | 2,846,164–2,886,164 | 1 | 2,866,164 | 4.28 × 10⁻6 | CRYBA4, CRYBB1, PLA2G1B, TPST2 |
| 56 | 1,992,379–2,032,379 | 1 | 2,012,379 | 4.64 × 10⁻6 | NYAP1 |
| 8 | 25,489,572–25,529,572 | 1 | 25,509,572 | 4.83 × 10⁻6 | CCKBR |
| Chr | SNP_R (bp) | SNP_N | Position_Top (bp) | p Value_Top | Candidate Gene |
|---|---|---|---|---|---|
| 22 | 2,458,265–2,498,265 | 1 | 2,478,265 | 4.23 × 10⁻8 | OGFOD1 |
| 3 | 16,905,242–16,945,242 | 1 | 16,925,242 | 7.84 × 10⁻8 | NFX1, OTULIN |
| 7 | 63,424,745–63,464,745 | 2 | 63,444,808 | 1.96 × 10⁻7 | ALDH18A1, CRYGB, ENTPD1 |
| 2 | 19,438,473–19,478,473 | 1 | 19,458,473 | 5.26 × 10⁻7 | SRFBP1 |
| 6 | 51,347,092–51,387,092 | 1 | 51,367,092 | 8.82 × 10⁻7 | CNRIP1, PLEK |
| 19 | 28,444,118–28,484,118 | 1 | 28,464,118 | 9.64 × 10⁻7 | HIC2 |
| 21 | 12,773,362–12,813,362 | 1 | 12,793,362 | 1.26 × 10⁻6 | ZFYVE20 |
| 1 | 85,267,483–85,307,483 | 1 | 85,287,483 | 1.54 × 10⁻6 | HCN1 |
| 7 | 10,056,229–10,096,229 | 1 | 10,076,229 | 2.42 × 10⁻6 | GPR85 |
| 25 | 24,937,562–24,977,562 | 1 | 24,957,562 | 2.45 × 10⁻6 | CDK16 |
| 6 | 60,924,314–60,964,314 | 1 | 60,944,314 | 2.96 × 10⁻6 | FNDC4 |
| 3 | 17,152,507–17,192,507 | 1 | 17,172,507 | 3.43 × 10⁻6 | INHBA |
| 1 | 87,476,833–87,516,833 | 1 | 87,496,833 | 3.57 × 10⁻6 | NARS |
| 51 | 4,080,177–4,120,177 | 1 | 4,100,177 | 3.66 × 10⁻6 | PLXNB3 |
| 20 | 13,829,146–13,869,146 | 1 | 13,849,146 | 4.07 × 10⁻6 | TMEM164 |
| 5 | 9,376,351–9,416,351 | 1 | 9,396,351 | 4.23 × 10⁻6 | FBXL4 |
| 36 | 405,705–445,705 | 1 | 425,705 | 4.64 × 10⁻6 | APOBEC3G |
| 1 | 53,107,797–53,147,797 | 1 | 53,127,797 | 4.96 × 10⁻6 | IQCM |
| Chr | SNP_R (bp) | SNP_N | Position_Top (bp) | p Value_Top | Candidate Gene |
|---|---|---|---|---|---|
| 12 | 32,000,256–32,040,256 | 5 | 32,035,778 | 3.54 × 10⁻8 | BAZ2B |
| 12 | 32,955,799–32,995,799 | 4 | 32,979,846 | 1.95 × 10⁻7 | ARL6IP6, PRPF40A |
| 12 | 32,292,886–32,332,886 | 6 | 32,331,576 | 1.96 × 10⁻7 | ACVR1, UPP2 |
| 12 | 33,242,523–33,282,523 | 5 | 33,281,526 | 1.97 × 10⁻7 | KALRN |
| 12 | 33,735,020–33,775,020 | 3 | 33,755,020 | 2.83 × 10⁻7 | GMPPA, PNKD |
| 12 | 33,222,041–33,262,041 | 6 | 33,256,286 | 2.90 × 10⁻7 | KALRN |
| 1 | 90,268,253–90,308,253 | 1 | 90,288,253 | 2.93 × 10⁻7 | HTR4 |
| 12 | 32,383,120–32,423,120 | 10 | 32,411,784 | 3.09 × 10⁻7 | CYTIP, ERMN, GALNT5 |
| 12 | 32,313,856–32,353,856 | 5 | 32,333,856 | 4.12 × 10⁻7 | ACVR1 |
| 12 | 33,088,046–33,128,046 | 3 | 33,126,288 | 4.89 × 10⁻7 | MYLK |
| 12 | 32,359,131–32,399,131 | 4 | 32,379,835 | 5.00 × 10⁻7 | CYTIP |
| 12 | 32,989,267–33,029,267 | 3 | 33,009,267 | 5.21 × 10⁻7 | fmnl2 |
| 12 | 33,759,093–33,799,093 | 3 | 33,779,093 | 5.90 × 10⁻7 | DARS, MCM6, PNKD, TMBIM1 |
| 10 | 44,336,019–44,376,019 | 1 | 44,356,019 | 6.05 × 10⁻7 | INSIG2 |
| 12 | 35,795,634–35,835,634 | 2 | 35,832,970 | 7.09 × 10⁻7 | LYPD6 |
| 12 | 32,500,094–32,540,094 | 2 | 32,520,094 | 7.64 × 10⁻7 | GPD2 |
| 12 | 32,930,958–32,970,958 | 6 | 32,959,646 | 7.85 × 10⁻7 | ARL6IP6 |
| 12 | 32,849,926–32,889,926 | 3 | 32,881,001 | 8.75 × 10⁻7 | GALNT13 |
| 12 | 32,870,402–32,910,402 | 3 | 32,890,482 | 9.58 × 10⁻7 | RPRM |
| 12 | 32,520,728–32,560,728 | 2 | 32,541,704 | 1.01 × 10⁻6 | NR4A2 |
| 17 | 4,638,093–4,678,093 | 1 | 4,658,093 | 1.10 × 10⁻6 | ZNF536 |
| 12 | 33,874,939–33,914,939 | 7 | 33,894,939 | 1.11 × 10⁻6 | THSD7B |
| 12 | 36,126,431–36,166,431 | 1 | 36,146,431 | 1.12 × 10⁻6 | UBXN4, enc |
| 12 | 34,918,833–34,958,833 | 4 | 34,956,534 | 1.20 × 10⁻6 | GTDC1 |
| 16 | 20,101,682–20,141,682 | 1 | 20,121,682 | 1.23 × 10⁻6 | DNAJC17 |
| 12 | 32,335,824–32,375,824 | 4 | 32,363,598 | 1.27 × 10⁻6 | ACVR1C |
| 15 | 11,074,569–11,114,569 | 1 | 11,094,569 | 1.32 × 10⁻6 | ADAP1, COX19 |
| 12 | 31,713,257–31,753,257 | 2 | 31,747,294 | 1.44 × 10⁻6 | TBR1 |
| 12 | 31,739,419–31,779,419 | 1 | 31,759,419 | 1.63 × 10⁻6 | PSMD14, TBR1 |
| 12 | 33,184,361–33,224,361 | 3 | 33,213,805 | 1.79 × 10⁻6 | KALRN, ROPN1 |
| 12 | 32,082,038–32,122,038 | 2 | 32,117,571 | 1.82 × 10⁻6 | TANC1 |
| 12 | 31,495,949–31,535,949 | 2 | 31,527,074 | 1.83 × 10⁻6 | KCNH7 |
| 45 | 2,422,442–2,462,442 | 1 | 2,442,442 | 1.90 × 10⁻6 | RNF39, ZKSCAN8 |
| 56 | 338,814–378,814 | 1 | 358,814 | 2.14 × 10⁻6 | BCL6B, SLC16A13 |
| 12 | 32,031,529–32,071,529 | 3 | 32,051,529 | 2.14 × 10⁻6 | WDSUB1 |
| 4 | 29,653,490–29,693,490 | 1 | 29,673,490 | 2.19 × 10⁻6 | XKR4 |
| 12 | 36,422,230–36,462,230 | 1 | 36,442,230 | 2.20 × 10⁻6 | ESYT3, FAIM |
| 12 | 32,158,497–32,198,497 | 5 | 32,181,649 | 2.21 × 10⁻6 | TANC1 |
| 4 | 19,975,042–20,015,042 | 1 | 19,995,042 | 2.29 × 10⁻6 | TOP1MT |
| 1 | 112,313,498–112,353,498 | 1 | 112,333,498 | 2.33 × 10⁻6 | ARID3C |
| 37 | 1,687,436–1,727,436 | 1 | 1,707,436 | 2.35 × 10⁻6 | AMIGO1, CYB561D1 |
| 17 | 30,419,784–30,459,784 | 1 | 30,439,784 | 2.47 × 10⁻6 | GALR2 |
| 21 | 24,163,402–24,203,402 | 1 | 24,183,402 | 2.48 × 10⁻6 | AKAP14, NDUFA1, NKAP, RPL39, SOWAHD, UPF3B |
| 12 | 36,200,620–36,240,620 | 1 | 36,220,620 | 2.57 × 10⁻6 | MAP3K19, RAB3GAP1 |
| 12 | 34,895,887–34,935,887 | 3 | 34,932,449 | 2.70 × 10⁻6 | GTDC1 |
| 12 | 32,262,572–32,302,572 | 2 | 32,299,191 | 2.86 × 10⁻6 | CCDC148, UPP2 |
| 12 | 36,763,187–36,803,187 | 1 | 36,783,187 | 2.90 × 10⁻6 | DDX18, Htr5b |
| 12 | 35,557,188–35,597,188 | 1 | 35,577,188 | 3.02 × 10⁻6 | ACVR2A |
| 12 | 34,667,954–34,707,954 | 1 | 34,687,954 | 3.04 × 10⁻6 | KYNU |
| 12 | 32,780,874–32,820,874 | 1 | 32,800,874 | 3.09 × 10⁻6 | KCNJ3 |
| 12 | 32,198,180–32,238,180 | 2 | 32,218,180 | 3.11 × 10⁻6 | PKP4, dapl1 |
| 12 | 31,634,734–31,674,734 | 1 | 31,654,734 | 3.19 × 10⁻6 | ADCY10, GCG |
| 6 | 79,213,869–79,253,869 | 1 | 79,233,869 | 3.33 × 10⁻6 | MARCKS |
| 12 | 31,950,349–31,990,349 | 1 | 31,970,349 | 3.44 × 10⁻6 | MARCH7 |
| 12 | 36,500,960–36,540,960 | 1 | 36,520,960 | 3.54 × 10⁻6 | C2orf76, DBI, STEAP3, TMEM37 |
| 12 | 34,988,988–35,028,988 | 1 | 35,008,988 | 3.69 × 10⁻6 | ZEB2 |
| 4 | 34,744,439–34,784,439 | 1 | 34,764,439 | 3.75 × 10⁻6 | B4GALT6, TTR |
| 41 | 5,614,428–5,654,428 | 1 | 5,634,428 | 3.82 × 10⁻6 | INA |
| 12 | 33,800,017–33,840,017 | 1 | 33,820,017 | 4.56 × 10⁻6 | CXCR4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, H.; Dong, T.; Wang, W.; Yan, X.; Geng, C.; Bai, S.; Hu, H. GWAS Enhances Genomic Prediction Accuracy of Caviar Yield, Caviar Color and Body Weight Traits in Sturgeons Using Whole-Genome Sequencing Data. Int. J. Mol. Sci. 2024, 25, 9756. https://doi.org/10.3390/ijms25179756
Song H, Dong T, Wang W, Yan X, Geng C, Bai S, Hu H. GWAS Enhances Genomic Prediction Accuracy of Caviar Yield, Caviar Color and Body Weight Traits in Sturgeons Using Whole-Genome Sequencing Data. International Journal of Molecular Sciences. 2024; 25(17):9756. https://doi.org/10.3390/ijms25179756
Chicago/Turabian StyleSong, Hailiang, Tian Dong, Wei Wang, Xiaoyu Yan, Chenfan Geng, Song Bai, and Hongxia Hu. 2024. "GWAS Enhances Genomic Prediction Accuracy of Caviar Yield, Caviar Color and Body Weight Traits in Sturgeons Using Whole-Genome Sequencing Data" International Journal of Molecular Sciences 25, no. 17: 9756. https://doi.org/10.3390/ijms25179756