
How Protein Depletion Balances Thrombosis and Bleeding Risk in the Context of Platelet’s Activatory and Negative Signaling

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
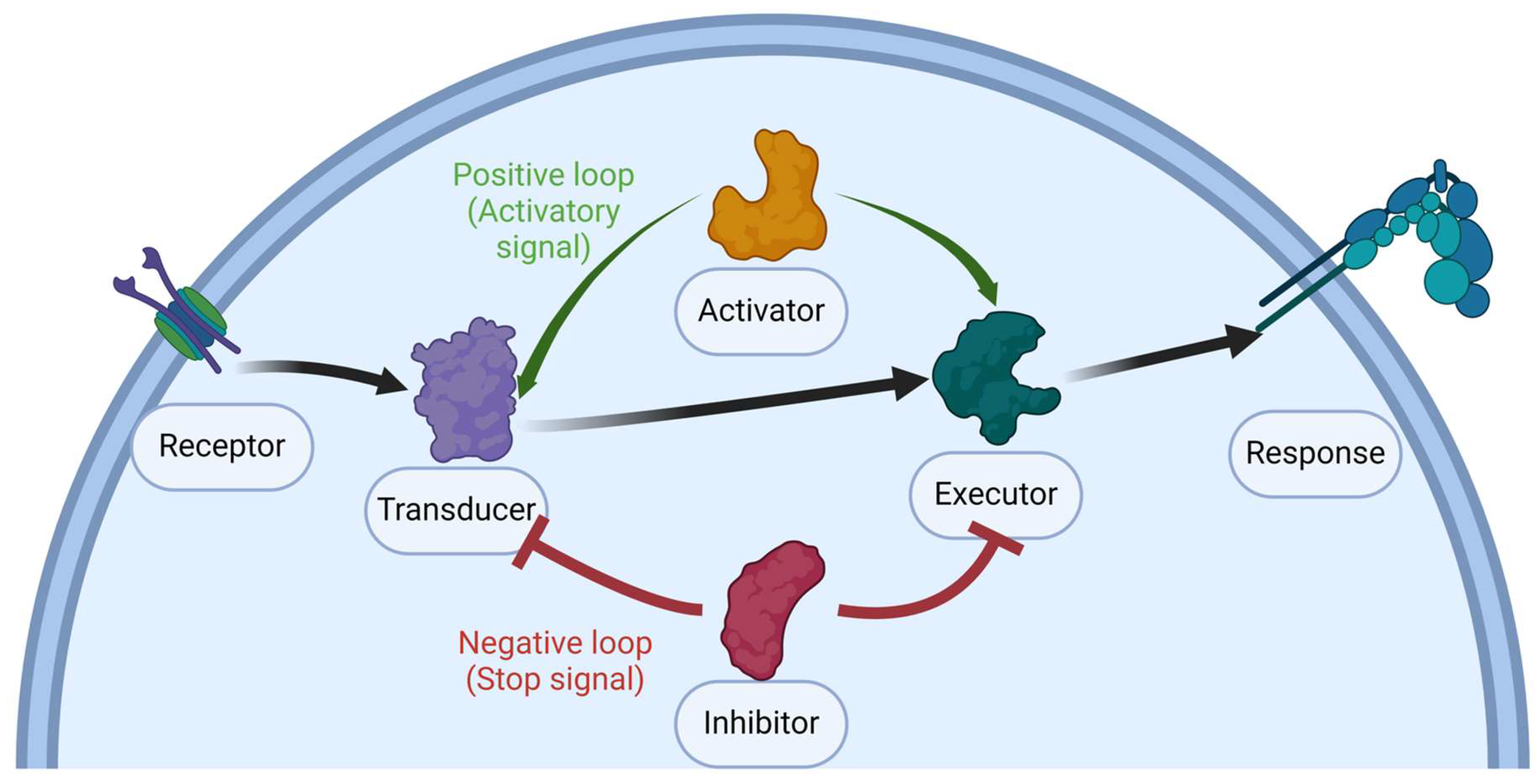
2. A Simple Overview of the Activation Process
3. Balance in the Pathways
4. Proteins
4.1. Integrins and Integrin-Associated Proteins
4.2. G Protein-Coupled Receptors (GPCRs)
4.3. Spreading and Cytoskeleton Dynamics
4.4. Platelet Secretion
5. Inhibitory Compounds with Effect on the Bleeding Time
6. Proteins without a Significative Difference in Bleeding Time or Thrombus Formation
7. Clinical Relevance
8. Future of Platelet Signaling and the Pathway Forward
9. Future Considerations
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Glossary
| AKT | Protein Kinase B |
| ASK1 | Apoptosis Signal-regulating Kinase 1 |
| C3 | Complement component 3 |
| Ca2+ | Calcium |
| CD148 | Protein tyrosine phosphatase receptor type J |
| CD154 | CD40 ligand |
| CD40 | Tumor necrosis factor receptor superfamily member 5 |
| CIB1 | Calcium and integrin-binding protein 1 |
| CLP36 | PDZ and LIM domain protein 1 |
| DASA | Diarylsulfonamide |
| DCa | Dichloroacetate |
| DHEA | Dehydroepiandrosterone sulphate |
| Dok-1 | Docking protein 1 |
| Dok-2 | Docking protein 2 |
| ELMO1 | Engulfment and cell motility protein 1 |
| ERK | Mitogen-activated protein kinase 15 |
| ERp57 | Protein disulfide-isomerase A3 |
| FlnA | Filamin A |
| Fyn | Tyrosine-protein kinase Fyn |
| GDF-15 | Growth/differentiation factor 15 |
| Gi | Guanine nucleotide-binding protein G(i) |
| GPCRs | G protein-coupled receptors |
| GPIb | Platelet glycoprotein Ib |
| GPV | Platelet glycoprotein V |
| GPIX | Platelet glycoprotein IX |
| GPVI | Platelet glycoprotein VI |
| GRIP1 | Glutamate receptor-interacting protein 1 |
| GSK3β | Glycogen synthase kinase-3 beta |
| Gα(i2) | Guanine nucleotide-binding protein G(i2) subunit alpha |
| Gα13 | Guanine nucleotide-binding protein G(13) subunit alpha-1 |
| HMGB1 | High mobility group protein B1 |
| IKKβ | Inhibitor of nuclear factor kappa-B kinase subunit beta |
| ILK | Integrin-linked protein kinase |
| iPLA2γ | Calcium-independent phospholipase A2-gamma |
| IκB | NF-kappa-B inhibitor alpha |
| JNK1 | Mitogen-activated protein kinase 8 |
| KIND3 | Fermitin family homolog 3 |
| M3BIM | Benzimidazole-derived oligosaccharide |
| MEK | Mitogen-activated protein kinase kinase |
| Munc18-2 | Mammalian Uncoordinated-18 |
| P2X1 | P2X purinoceptor 1 |
| P2Y12 | P2Y purinoceptor 12 |
| p38 MAPK | Mitogen-activated protein kinase 11 |
| Panx | Pannexin-1 |
| PAR1 | Protease-activated receptor-1 |
| PDK1 | 3-Phosphoinositide-dependent protein kinase 1 |
| PECAM-1 | Platelet and Endothelial Cell Adhesion Molecule 1 |
| PI3K | Phosphatidylinositol 45-bisphosphate 3-kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PP2B | Serine/threonine-protein phosphatase 2B |
| PTEN | Phosphatase and tensin homolog |
| RAC1 | Ras-related C3 botulinum toxin substrate 1 |
| Raf1 | Proto-oncogene c-RAF |
| Rap1 | Ras-related protein 1 |
| RGS 16 | Regulator of G protein signaling 16 |
| RhoA | Ras homolog family member A |
| RhoG | Ras homolog family member G |
| RIAM | Rap1–GTP-interacting adapter molecule |
| RIP3 | Receptor-interacting protein kinase 3 |
| ROCK | Rho-associated protein kinase |
| SERCA3 | Sarcoplasmic/endoplasmic reticulum Calcium ATPase 3 |
| SFK | Src kinase family |
| Shp2 | Src homology region 2-containing protein tyrosine phosphatase 2 |
| SNARE | SNAP receptors |
| Src | Proto-oncogene tyrosine-protein kinase Src |
| TAIII | Timosaponin A-III |
| TLR4/cGKI | Toll-like receptor 4 |
| TMX1 | Thioredoxin-related Transmembrane Protein 1 |
| TNFR | Tumor necrosis factor receptors |
| TP receptor | Thromboxane receptor |
| TRAF3 | TNF receptor-associated factor 3 |
| TSSC6 | Tumor-suppressing STF cDNA 6 |
| TXA2 | Thromboxane A2 |
| VPS33B | Vacuolar protein sorting-associated protein 33B |
References
- Boudreaux, M.K.; Christopherson, P.W. Platelet Structure. In Schalm’s Veterinary Hematology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2022; pp. 658–666. [Google Scholar] [CrossRef]
- Yadav, S.; Storrie, B. The cellular basis of platelet secretion: Emerging structure/function relationships. Platelets 2017, 28, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Yi, H.S.; Kim, Y.; Kroh, E.M.; Chien, J.W.; Eaton, K.D.; Goodman, M.T.; Tait, J.F.; Tewari, M.; Pritchard, C.C. Plasma processing conditions substantially influence circulating microRNA biomarker levels. PLoS ONE 2013, 8, e64795. [Google Scholar] [CrossRef]
- Periayah, M.H.; Halim, A.S.; Saad, A.Z.M. Mechanism action of platelets and crucial blood coagulation pathways in Hemostasis. Int. J. Hematol.-Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar] [PubMed]
- Parker, W.A.E.; Storey, R.F. The role of platelet P2Y(12) receptors in inflammation. Br. J. Pharmacol. 2023, 181, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Belyaev, A.V.; Kushchenko, Y.K. Biomechanical activation of blood platelets via adhesion to von Willebrand factor studied with mesoscopic simulations. Biomech. Model. Mechanobiol. 2023, 22, 785–808. [Google Scholar] [CrossRef] [PubMed]
- Graca, F.A.; Minden-Birkenmaier, B.A.; Stephan, A.; Demontis, F.; Labelle, M. Signaling roles of platelets in skeletal muscle regeneration. BioEssays 2023, 45, e2300134. [Google Scholar] [CrossRef]
- Horev, M.B.; Zabary, Y.; Zarka, R.; Sorrentino, S.; Medalia, O.; Zaritsky, A.; Geiger, B. Differential dynamics of early stages of platelet adhesion and spreading on collagen IV- and fibrinogen-coated surfaces. F1000Research 2020, 9, 449. [Google Scholar] [CrossRef]
- Mitchell, J.L.; Little, G.; Bye, A.P.; Gaspar, R.S.; Unsworth, A.J.; Kriek, N.; Sage, T.; Stainer, A.; Sangowawa, I.; Morrow, G.B.; et al. Platelet factor XIII-A regulates platelet function and promotes clot retraction and stability. Res. Pract. Thromb. Haemost. 2023, 7, 100200. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Kim, S.; Kim, S. An Insight into Recent Advances on Platelet Function in Health and Disease. Int. J. Mol. Sci. 2022, 23, 6022. [Google Scholar] [CrossRef]
- Warny, M.; Helby, J.; Birgens, H.S.; Bojesen, S.E.; Nordestgaard, B.G. Arterial and venous thrombosis by high platelet count and high hematocrit: 108,521 individuals from the Copenhagen General Population Study. J. Thromb. Haemost. 2019, 17, 1898–1911. [Google Scholar] [CrossRef]
- Rawish, E.; Nording, H.; Münte, T.; Langer, H.F. Platelets as Mediators of Neuroinflammation and Thrombosis. Front. Immunol. 2020, 11, 548631. [Google Scholar] [CrossRef]
- Michael, C.; Pancaldi, F.; Britton, S.; Kim, O.V.; Peshkova, A.D.; Vo, K.; Xu, Z.; Litvinov, R.I.; Weisel, J.W.; Alber, M. Combined computational modeling and experimental study of the biomechanical mechanisms of platelet-driven contraction of fibrin clots. Commun. Biol. 2023, 6, 869. [Google Scholar] [CrossRef]
- Chen, C.; Li, Y.; Yu, Z.; Liu, Z.; Shi, Y.; Lewandowska, U.; Sobczak, M.; Fichna, J.; Kreis, M. Platelet activity in the pathophysiology of inflammatory bowel diseases. Curr. Drug Targets 2015, 16, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Rondina, M.T.; Carlisle, M.; Fraughton, T.; Brown, S.M.; Miller, R.R., 3rd; Harris, E.S.; Weyrich, A.S.; Zimmerman, G.A.; Supiano, M.A.; Grissom, C.K. Platelet-monocyte aggregate formation and mortality risk in older patients with severe sepsis and septic shock. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 225–231. [Google Scholar] [CrossRef]
- Lood, C.; Tydén, H.; Gullstrand, B.; Klint, C.; Wenglén, C.; Nielsen, C.T.; Heegaard, N.H.; Jönsen, A.; Kahn, R.; Bengtsson, A.A. Type I interferon-mediated skewing of the serotonin synthesis is associated with severe disease in systemic lupus erythematosus. PLoS ONE 2015, 10, e0125109. [Google Scholar] [CrossRef]
- Miller, E.A.; Gopal, R.; Valdes, V.; Berger, J.S.; Bhardwaj, N.; O’Brien, M.P. Soluble CD40 ligand contributes to dendritic cell-mediated T-cell dysfunction in HIV-1 infection. AIDS 2015, 29, 1287–1296. [Google Scholar] [CrossRef]
- Chew, D.P.; Bhatt, D.L.; Sapp, S.; Topol, E.J. Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists: A meta-analysis of phase III multicenter randomized trials. Circulation 2001, 103, 201–206. [Google Scholar] [CrossRef]
- Estevez, B.; Shen, B.; Du, X. Targeting integrin and integrin signaling in treating thrombosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 24–29. [Google Scholar] [CrossRef]
- Sakuma, T.; Sari, I.; Goodman, C.N.; Lindner, J.R.; Klibanov, A.L.; Kaul, S. Simultaneous integrin alphavbeta3 and glycoprotein IIb/IIIa inhibition causes reduction in infarct size in a model of acute coronary thrombosis and primary angioplasty. Cardiovasc. Res. 2005, 66, 552–561. [Google Scholar] [CrossRef]
- Munksgaard Thorén, M.; Chmielarska Masoumi, K.; Krona, C.; Huang, X.; Kundu, S.; Schmidt, L.; Forsberg-Nilsson, K.; Floyd Keep, M.; Englund, E.; Nelander, S.; et al. Integrin α10, a Novel Therapeutic Target in Glioblastoma, Regulates Cell Migration, Proliferation, and Survival. Cancers 2019, 11, 587. [Google Scholar] [CrossRef]
- Ahmad, K.; Lee, E.J.; Shaikh, S.; Kumar, A.; Rao, K.M.; Park, S.Y.; Jin, J.O.; Han, S.S.; Choi, I. Targeting integrins for cancer management using nanotherapeutic approaches: Recent advances and challenges. Semin. Cancer Biol. 2021, 69, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Xu, Y.; Wang, Y.; Kumar, A.; Peters, D.M.; Du, Y. α5β1 Integrin Promotes Anchoring and Integration of Transplanted Stem Cells to the Trabecular Meshwork in the Eye for Regeneration. Stem Cells Dev. 2020, 29, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Tresoldi, I.; Sangiuolo, C.F.; Manzari, V.; Modesti, A. SARS-COV-2 and infectivity: Possible increase in infectivity associated to integrin motif expression. J. Med. Virol. 2020, 92, 1741–1742. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.H.; Holubar, S.; Rieder, F. Fibrostenotic strictures in Crohn’s disease. Intest. Res. 2020, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Phanthaphol, N.; Somboonpatarakun, C.; Suwanchiwasiri, K.; Chieochansin, T.; Sujjitjoon, J.; Wongkham, S.; Maher, J.; Junking, M.; Yenchitsomanus, P.T. Chimeric Antigen Receptor T Cells Targeting Integrin αvβ6 Expressed on Cholangiocarcinoma Cells. Front. Oncol. 2021, 11, 657868. [Google Scholar] [CrossRef]
- Park, E.J.; Myint, P.K.; Appiah, M.G.; Darkwah, S.; Caidengbate, S.; Ito, A.; Matsuo, E.; Kawamoto, E.; Gaowa, A.; Shimaoka, M. The Spike Glycoprotein of SARS-CoV-2 Binds to β1 Integrins Expressed on the Surface of Lung Epithelial Cells. Viruses 2021, 13, 645. [Google Scholar] [CrossRef]
- Mrugacz, M.; Bryl, A.; Falkowski, M.; Zorena, K. Integrins: An Important Link between Angiogenesis, Inflammation and Eye Diseases. Cells 2021, 10, 1703. [Google Scholar] [CrossRef]
- Ho, T.C.; Yeh, S.I.; Chen, S.L.; Tsao, Y.P. Integrin αv and Vitronectin Prime Macrophage-Related Inflammation and Contribute the Development of Dry Eye Disease. Int. J. Mol. Sci. 2021, 22, 8410. [Google Scholar] [CrossRef]
- Beaudoin, C.A.; Hamaia, S.W.; Huang, C.L.; Blundell, T.L.; Jackson, A.P. Can the SARS-CoV-2 Spike Protein Bind Integrins Independent of the RGD Sequence? Front. Cell. Infect. Microbiol. 2021, 11, 765300. [Google Scholar] [CrossRef]
- Stojanovic, A.; Hwa, J. Rhodopsin and retinitis pigmentosa: Shedding light on structure and function. Recept. Channels 2002, 8, 33–50. [Google Scholar]
- Wu, A.; Salom, D.; Hong, J.; Pardon, E.; Steyaert, J.; Kiser, P.D.; Palczewski, K. Structure-guided GPCR drug development of human single domain camelid antibodies (Nanobody) for autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2023, 64, 765. [Google Scholar]
- Deen, P.M.; Marr, N.; Kamsteeg, E.-J.; van Balkom, B.W. Nephrogenic diabetes insipidus. Curr. Opin. Nephrol. Hypertens. 2000, 9, 591–595. [Google Scholar] [PubMed]
- Heifetz, A.; Barker, O.; Verquin, G.; Wimmer, N.; Meutermans, W.; Pal, S.; Law, R.J.; Whittaker, M. Fighting obesity with a sugar-based library: Discovery of novel MCH-1R antagonists by a new computational–VAST approach for exploration of GPCR binding sites. J. Chem. Inf. Model. 2013, 53, 1084–1099. [Google Scholar] [PubMed]
- Kimple, M.E.; Neuman, J.C.; Linnemann, A.K.; Casey, P.J. Inhibitory G proteins and their receptors: Emerging therapeutic targets for obesity and diabetes. Exp. Mol. Med. 2014, 46, e102. [Google Scholar]
- Liu, T.; Ji, R.-L.; Tao, Y.-X. Naturally occurring mutations in G protein-coupled receptors associated with obesity and type 2 diabetes mellitus. Pharmacol. Ther. 2022, 234, 108044. [Google Scholar]
- Capote, L.A.; Perez, R.M.; Lymperopoulos, A.J. GPCR signaling and cardiac function. Eur. J. Pharmacol. 2015, 763, 143–148. [Google Scholar]
- Brinks, H.L.; Eckhart, A.D. Regulation of GPCR signaling in hypertension. Biochim. Biophys. Acta 2010, 1802, 1268–1275. [Google Scholar]
- Deshpande, D.A.; Penn, R.B. Targeting G protein-coupled receptor signaling in asthma. Cell. Signal. 2006, 18, 2105–2120. [Google Scholar]
- Wendell, S.G.; Fan, H.; Zhang, C. G protein–coupled receptors in asthma therapy: Pharmacology and drug action. Pharmacol. Rev. 2020, 72, 1–49. [Google Scholar]
- Wright, D.; Tripathi, S.; Sikarwar, A.; Santosh, K.; Perez-Zoghbi, J.; Ojo, O.; Irechukwu, N.; Ward, J.; Schaafsma, D. Regulation of GPCR-mediated smooth muscle contraction: Implications for asthma and pulmonary hypertension. Pulm. Pharmacol. Ther. 2013, 26, 121–131. [Google Scholar]
- García, M.; González de Buitrago, J.; Jiménez-Rosés, M.; Pardo, L.; Hinkle, P.M.; Moreno, J.C. Central hypothyroidism due to a TRHR mutation causing impaired ligand affinity and transactivation of Gq. J. Clin. Endocrinol. Metab. 2017, 102, 2433–2442. [Google Scholar] [PubMed]
- Babwah, A.V. The wonderful and masterful G protein-coupled receptor (GPCR): A focus on signaling mechanisms and the neuroendocrine control of fertility. Mol. Cell. Endocrinol. 2020, 515, 110886. [Google Scholar] [PubMed]
- Zhang, D.; Wang, Y.; Lin, H.; Sun, Y.; Wang, M.; Jia, Y.; Yu, X.; Jiang, H.; Xu, W.; Sun, J.; et al. Function and therapeutic potential of G protein-coupled receptors in epididymis. Br. J. Pharmacol. 2020, 177, 5489–5508. [Google Scholar] [PubMed]
- Peng, W.-T.; Sun, W.-Y.; Li, X.-R.; Sun, J.-C.; Du, J.-J.; Wei, W. Emerging roles of G protein-coupled receptors in hepatocellular carcinoma. Int. J. Mol. Sci. 2018, 19, 1366. [Google Scholar] [CrossRef] [PubMed]
- Gad, A.A.; Balenga, N. The emerging role of adhesion GPCRs in cancer. ACS Pharmacol. Transl. Sci. 2020, 3, 29–42. [Google Scholar]
- Zhu, X.; Lee, H.G.; Raina, A.K.; Perry, G.; Smith, M.A. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 2002, 11, 270–281. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Z.; Sittirattanayeungyong, S.; Hongpaisan, J. ApoE4-related microvascular disease in the Alzheimer’s disease hippocampal CA1 stratum radiatum. Neuroscience 2023, 526, 204–222. [Google Scholar] [CrossRef]
- Elango, R.; Banaganapalli, B.; Mujalli, A.; AlRayes, N.; Almaghrabi, S.; Almansouri, M.; Sahly, A.; Jadkarim, G.A.; Malik, M.Z.; Kutbi, H.I.; et al. Potential Biomarkers for Parkinson Disease from Functional Enrichment and Bioinformatic Analysis of Global Gene Expression Patterns of Blood and Substantia Nigra Tissues. Bioinform. Biol. Insights 2023, 17, 11779322231166214. [Google Scholar] [CrossRef]
- Iba, M.; Kim, C.; Kwon, S.; Szabo, M.; Horan-Portelance, L.; Peer, C.J.; Figg, W.D.; Reed, X.; Ding, J.; Lee, S.J.; et al. Inhibition of p38α MAPK restores neuronal p38γ MAPK and ameliorates synaptic degeneration in a mouse model of DLB/PD. Sci. Transl. Med. 2023, 15, eabq6089. [Google Scholar] [CrossRef]
- Liu, H.; Ma, H.; Zeng, X.; Wu, C.; Acharya, S.; Sudan, S.K.; Zhang, X. Ubiquitination of GRK2 Is Required for the β-Arrestin-Biased Signaling Pathway of Dopamine D2 Receptors to Activate ERK Kinases. Int. J. Mol. Sci. 2023, 24, 10031. [Google Scholar] [CrossRef]
- Yue, W.; Deng, X.; Wang, Z.; Jiang, M.; Hu, R.; Duan, Y.; Wang, Q.; Cui, J.; Fang, Y. Inhibition of the MEK/ERK pathway suppresses immune overactivation and mitigates TDP-43 toxicity in a Drosophila model of ALS. Immun. Ageing 2023, 20, 27. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Upadhayay, S.; Mehan, S. Inhibition of extracellular regulated kinase (ERK)-1/2 signaling pathway in the prevention of ALS: Target inhibitors and influences on neurological dysfunctions. Eur. J. Cell Biol. 2021, 100, 151179. [Google Scholar] [CrossRef]
- Pérez-Cabello, J.A.; Silvera-Carrasco, L.; Franco, J.M.; Capilla-González, V.; Armaos, A.; Gómez-Lima, M.; García-García, R.; Yap, X.W.; Leal-Lasarte, M.; Lall, D.; et al. MAPK/MAK/MRK overlapping kinase (MOK) controls microglial inflammatory/type-I IFN responses via Brd4 and is involved in ALS. Proc. Natl. Acad. Sci. USA 2023, 120, e2302143120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shen, Q.; Wang, Y.; Deng, X.; Fan, J.; Gu, X.; Fan, M.; Wei, K.; Cheng, C.R.; Zhang, W.D.; et al. Corylifol A ameliorates muscle atrophy by inhibiting TAOK1/p38-MAPK/FoxO3 pathway in cancer cachexia. J. Cachexia Sarcopenia Muscle 2023, 14, 2098–2113. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Du, B.; Xi, X. Colorectal cancer cells induce the formation of cancer-associated fibroblasts by activating the ERK signaling pathway in fibroblasts. Nan Fang Yi Ke Da Xue Xue Bao J. South. Med. Univ. 2023, 43, 943–951. [Google Scholar] [CrossRef]
- Ma, W.; Wu, Z.; Maghsoudloo, M.; Ijaz, I.; Dehghan Shasaltaneh, M.; Zhang, Y.; Weng, Q.; Fu, J.; Imani, S.; Wen, Q.L. Dermokine mutations contribute to epithelial-mesenchymal transition and advanced melanoma through ERK/MAPK pathways. PLoS ONE 2023, 18, e0285806. [Google Scholar] [CrossRef]
- Rej, A.; Paladhi, A.; Daripa, S.; Sarkar, D.; Bhattacharyya, S.; Mondal, I.; Hira, S.K. Galunisertib synergistically potentiates the doxorubicin-mediated antitumor effect and kickstarts the immune system against aggressive lymphoma. Int. Immunopharmacol. 2023, 114, 109521. [Google Scholar] [CrossRef]
- Choi, C.; Thi Thao Tran, N.; Van Ngu, T.; Park, S.W.; Song, M.S.; Kim, S.H.; Bae, Y.-U.; Ayudthaya, P.D.N.; Munir, J.; Kim, E.; et al. Promotion of tumor progression and cancer stemness by MUC15 in thyroid cancer via the GPCR/ERK and integrin-FAK signaling pathways. Oncogenesis 2018, 7, 85. [Google Scholar]
- Yoon, S.S.; Kwon, H.W.; Shin, J.H.; Rhee, M.H.; Park, C.E.; Lee, D.H. Anti-Thrombotic Effects of Artesunate through Regulation of cAMP and PI3K/MAPK Pathway on Human Platelets. Int. J. Mol. Sci. 2022, 23, 1586. [Google Scholar] [CrossRef]
- Glotfelty, E.J.; Tovar, Y.R.L.B.; Hsueh, S.C.; Tweedie, D.; Li, Y.; Harvey, B.K.; Hoffer, B.J.; Karlsson, T.E.; Olson, L.; Greig, N.H. The RhoA-ROCK1/ROCK2 Pathway Exacerbates Inflammatory Signaling in Immortalized and Primary Microglia. Cells 2023, 12, 1367. [Google Scholar] [CrossRef]
- Mani, S.; Jindal, D.; Chopra, H.; Jha, S.K.; Singh, S.K.; Ashraf, G.M.; Kamal, M.; Iqbal, D.; Chellappan, D.K.; Dey, A.; et al. ROCK2 inhibition: A futuristic approach for the management of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2022, 142, 104871. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Rodríguez-Perez, A.I.; Villar-Cheda, B.; Borrajo, A.; Dominguez-Meijide, A.; Guerra, M.J. Rho Kinase and Dopaminergic Degeneration: A Promising Therapeutic Target for Parkinson’s Disease. Neuroscientist 2015, 21, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.; Subramaniam, M.D.; Venkatesan, D.; Cho, S.-G.; Ryding, M.; Meyer, M.; Vellingiri, B. Role of RhoA-ROCK signaling in Parkinson’s disease. Eur. J. Pharmacol. 2021, 894, 173815. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Tang, Y.-G.; Wang, L.; He, W.; Pan, H.-H.; Nie, R.-R.; Can, Y. Role of Rho-mediated ROCK-Semaphorin3A signaling pathway in the pathogenesis of Parkinson’s disease in a mouse model. J. Neurol. Sci. 2016, 370, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-C.; Zhao, H.; Zhang, W.-Q.; Li, Y.-Q.; Ren, L.-Q. The role of the Rho/Rock signaling pathway in the pathogenesis of acute ischemic myocardial fibrosis in rat models. Exp. Ther. Med. 2013, 5, 1123–1128. [Google Scholar] [CrossRef]
- Huang, Z.; Nan, C.; Wang, H.; Su, Q.; Xue, W.; Chen, Y.; Shan, X.; Duan, J.; Chen, G.; Tao, W. Crocetin ester improves myocardial ischemia via Rho/ROCK/NF-κB pathway. Int. Immunopharmacol. 2016, 38, 186–193. [Google Scholar] [CrossRef]
- Manintveld, O.C.; Verdouw, P.D.; Duncker, D.J. The Risk of Rock. Am. J. Physiol. Circ. Physiol. 2007, 292, H2563–H2565. [Google Scholar] [CrossRef]
- Sawada, N.; Liao, J.K. Rho/Rho-Associated Coiled-Coil Forming Kinase Pathway as Therapeutic Targets for Statins in Atherosclerosis. Antioxid. Redox Signal. 2014, 20, 1251–1267. [Google Scholar] [CrossRef]
- Zhou, Q.; Gensch, C.; Liao, J.K. Rho-associated coiled-coil-forming kinases (ROCKs): Potential targets for the treatment of atherosclerosis and vascular disease. Trends Pharmacol. Sci. 2011, 32, 167–173. [Google Scholar] [CrossRef]
- Zhou, Q.; Liao, J.K. Rho Kinase: An Important Mediator of Atherosclerosis and Vascular Disease. Curr. Pharm. Des. 2009, 15, 3108–3115. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [PubMed]
- Ryan, J.J. Tyrosine kinase inhibitors in pulmonary vascular disease. JACC Basic Transl. Sci. 2016, 1, 684–686. [Google Scholar] [PubMed]
- Manouchehri, A.; Kanu, E.; Mauro, M.J.; Aday, A.W.; Lindner, J.R.; Moslehi, J. Tyrosine kinase inhibitors in leukemia and cardiovascular events: From mechanism to patient care. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 301–308. [Google Scholar] [PubMed]
- Gomez-Puerta, J.A.; Mócsai, A. Tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Curr. Top. Med. Chem. 2013, 13, 760–773. [Google Scholar] [PubMed]
- Kim, H.-J.; Yoon, H.-J.; Choi, J.-Y.; Lee, I.-K.; Kim, S.-Y. The tyrosine kinase inhibitor GNF-2 suppresses osteoclast formation and activity. J. Leukoc. Biol. 2014, 95, 337–345. [Google Scholar]
- Schweig, J.E.; Yao, H.; Beaulieu-Abdelahad, D.; Ait-Ghezala, G.; Mouzon, B.; Crawford, F.; Mullan, M.; Paris, D. Alzheimer’s disease pathological lesions activate the spleen tyrosine kinase. Acta Neuropathol. Commun. 2017, 5, 69. [Google Scholar]
- Nygaard, H.B.; van Dyck, C.H.; Strittmatter, S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 8. [Google Scholar]
- Depboylu, C.; Höllerhage, M.; Schnurrbusch, S.; Brundin, P.; Oertel, W.H.; Schrattenholz, A.; Höglinger, G.U. Neuregulin-1 receptor tyrosine kinase ErbB4 is upregulated in midbrain dopaminergic neurons in Parkinson disease. Neurosci. Lett. 2012, 531, 209–214. [Google Scholar]
- Gabrielsen, A.; Qiu, H.; Bäck, M.; Hamberg, M.; Hemdahl, A.-L.; Agardh, H.; Folkersen, L.; Swedenborg, J.; Hedin, U.; Paulsson-Berne, G. Thromboxane synthase expression and thromboxane A 2 production in the atherosclerotic lesion. J. Mol. Med. 2010, 88, 795–806. [Google Scholar]
- Alamri, A.K.; Ma, C.L.; Ryan, J.J. Novel Drugs for the Treatment of Pulmonary Arterial Hypertension: Where Are We Going? Drugs 2023, 83, 577–585. [Google Scholar] [CrossRef]
- Chen, H. Role of thromboxane A2 signaling in endothelium-dependent contractions of arteries. Prostaglandins Other Lipid Mediat. 2018, 134, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Rucker, D.; Dhamoon, A.S. Physiology, Thromboxane A2; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Zhang, Y.; Steinmetz-Späh, J.; Idborg, H.; Zhu, L.; Li, H.; Rao, H.; Chen, Z.; Guo, Z.; Hu, L.; Xu, C.; et al. Microsomal prostaglandin E synthase-1 inhibition prevents adverse cardiac remodelling after myocardial infarction in mice. Br. J. Pharmacol. 2023, 180, 1981–1998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, Y.; Xing, X.; Li, H.; Wang, X.; Zhang, H.; Wang, X.; Li, X.; Li, Y.; Wang, Q. Electroacupuncture Treats Myocardial Infarction by Influencing the Regulation of Substance P in the Neurovascular to Modulate PGI2/TXA2 Metabolic Homeostasis via PI3K/AKT Pathway: A Bioinformatics-Based Multiomics and Experimental Study. Comput. Math. Methods Med. 2022, 2022, 5367753. [Google Scholar] [CrossRef] [PubMed]
- Estevez, B.; Du, X. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef]
- Montague, S.J.; Lim, Y.J.; Lee, W.M.; Gardiner, E.E. Imaging Platelet Processes and Function-Current and Emerging Approaches for Imaging in vitro and in vivo. Front. Immunol. 2020, 11, 78. [Google Scholar] [CrossRef]
- Tomaiuolo, M.; Litvinov, R.I.; Weisel, J.W.; Stalker, T.J. Use of electron microscopy to study platelets and thrombi. Platelets 2020, 31, 580–588. [Google Scholar] [CrossRef]
- Alkarithi, G.; Duval, C.; Shi, Y.; Macrae, F.L.; Ariëns, R.A.S. Thrombus Structural Composition in Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2021, 41, 2370–2383. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, P.; Solari, F.A.; Sickmann, A.; Garcia, A.; Jurk, K.; Heemskerk, J.W.M. Molecular Proteomics and Signalling of Human Platelets in Health and Disease. Int. J. Mol. Sci. 2021, 22, 9860. [Google Scholar] [CrossRef]
- Seifert, J.; Rheinlaender, J.; Lang, F.; Gawaz, M.; Schäffer, T.E. Thrombin-induced cytoskeleton dynamics in spread human platelets observed with fast scanning ion conductance microscopy. Sci. Rep. 2017, 7, 4810. [Google Scholar] [CrossRef]
- Mwiza, J.M.N.; Lee, R.H.; Paul, D.S.; Holle, L.A.; Cooley, B.C.; Nieswandt, B.; Schug, W.J.; Kawano, T.; Mackman, N.; Wolberg, A.S.; et al. Both G protein-coupled and immunoreceptor tyrosine-based activation motif receptors mediate venous thrombosis in mice. Blood 2022, 139, 3194–3203. [Google Scholar] [CrossRef]
- Khatlani, T.; Pradhan, S.; Da, Q.; Gushiken, F.C.; Bergeron, A.L.; Langlois, K.W.; Molkentin, J.D.; Rumbaut, R.E.; Vijayan, K.V. The β isoform of the catalytic subunit of protein phosphatase 2B restrains platelet function by suppressing outside-in αII b β3 integrin signaling. J. Thromb. Haemost. 2014, 12, 2089–2101. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hong, J.; Zhong, H.; Zhao, Y.; Li, J.; Shen, W.; Luo, X.; Shi, H.; Hu, L.; Liu, J.; et al. IL-37 Attenuates Platelet Activation and Thrombosis Through IL-1R8 Pathway. Circ. Res. 2023, 132, e134–e150. [Google Scholar] [CrossRef] [PubMed]
- Bye, A.P.; Unsworth, A.J.; Gibbins, J.M. Platelet signaling: A complex interplay between inhibitory and activatory networks. J. Thromb. Haemost. 2016, 14, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, L.; Bergmeier, W. Negative regulators of platelet activation and adhesion. J. Thromb. Haemost. 2018, 16, 220–230. [Google Scholar] [CrossRef]
- Petzold, T.; Ruppert, R.; Pandey, D.; Barocke, V.; Meyer, H.; Lorenz, M.; Zhang, L.; Siess, W.; Massberg, S.; Moser, M. β1 integrin−mediated signals are required for platelet granule secretion and hemostasis in mouse. Blood 2013, 122, 2723–2731. [Google Scholar] [CrossRef]
- Niki, M.; Nayak, M.K.; Jin, H.; Bhasin, N.; Plow, E.F.; Pandolfi, P.P.; Rothman, P.B.; Chauhan, A.K.; Lentz, S.R. Dok-1 negatively regulates platelet integrin alphaIIbbeta3 outside-in signalling and inhibits thrombosis in mice. Thromb. Haemost. 2016, 115, 969–978. [Google Scholar] [CrossRef]
- Nieswandt, B.; Moser, M.; Pleines, I.; Varga-Szabo, D.; Monkley, S.; Critchley, D.; Fassler, R. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J. Exp. Med. 2007, 204, 3113–3118. [Google Scholar] [CrossRef]
- Petrich, B.G.; Marchese, P.; Ruggeri, Z.M.; Spiess, S.; Weichert, R.A.; Ye, F.; Tiedt, R.; Skoda, R.C.; Monkley, S.J.; Critchley, D.R.; et al. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J. Exp. Med. 2007, 204, 3103–3111. [Google Scholar] [CrossRef]
- Stefanini, L.; Ye, F.; Snider, A.K.; Sarabakhsh, K.; Piatt, R.; Paul, D.S.; Bergmeier, W.; Petrich, B.G. A talin mutant that impairs talin-integrin binding in platelets decelerates alphaIIbbeta3 activation without pathological bleeding. Blood 2014, 123, 2722–2731. [Google Scholar] [CrossRef]
- Lagarrigue, F.; Paul, D.S.; Gingras, A.R.; Valadez, A.J.; Sun, H.; Lin, J.; Cuevas, M.N.; Ablack, J.N.G.; Lopez Ramirez, M.A.; Bergmeier, W.; et al. Talin1 is the Principal Platelet Rap1 Effector of Integrin Activation. Blood 2020, 136, 1180–1190. [Google Scholar] [CrossRef]
- Tucker, K.L.; Sage, T.; Stevens, J.M.; Jordan, P.A.; Jones, S.; Barrett, N.E.; St-Arnaud, R.; Frampton, J.; Dedhar, S.; Gibbins, J.M. A dual role for integrin-linked kinase in platelets: Regulating integrin function and alpha-granule secretion. Blood 2008, 112, 4523–4531. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.I.; Tucker, K.L.; Sasikumar, P.; Sage, T.; Kaiser, W.J.; Moore, C.; Emerson, M.; Gibbins, J.M. Integrin-linked kinase regulates the rate of platelet activation and is essential for the formation of stable thrombi. J. Thromb. Haemost. 2014, 12, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Crow, A.R.; Leytin, V.; Starkey, A.F.; Rand, M.L.; Lazarus, A.H. CD154 (CD40 ligand)-deficient mice exhibit prolonged bleeding time and decreased shear-induced platelet aggregates. J. Thromb. Haemost. 2003, 1, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.U.; Patel, P.; Derstine, R.; Turaga, R.; Chen, X.; Golla, K.; Neeves, K.B.; Ichijo, H.; Naik, U.P. Ask1 regulates murine platelet granule secretion, thromboxane A2 generation, and thrombus formation. Blood 2017, 129, 1197–1209. [Google Scholar] [CrossRef]
- Sledz, K.M.; Moore, S.F.; Vijayaragavan, V.; Mallah, S.; Goudswaard, L.J.; Williams, C.M.; Hunter, R.W.; Hers, I. Redundant role of ASK1-mediated p38MAPK activation in human platelet function. Cell. Signal. 2020, 68, 109528. [Google Scholar] [CrossRef]
- Holbrook, L.M.; Sasikumar, P.; Stanley, R.G.; Simmonds, A.D.; Bicknell, A.B.; Gibbins, J.M. The platelet-surface thiol isomerase enzyme ERp57 modulates platelet function. J. Thromb. Haemost. 2012, 10, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ahmad, S.S.; Zhou, J.; Wang, L.; Cully, M.P.; Essex, D.W. The disulfide isomerase ERp57 mediates platelet aggregation, hemostasis, and thrombosis. Blood 2012, 119, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.; Zhou, J.; Ahmad, S.S.; Mutus, B.; Garbi, N.; Hammerling, G.; Liu, J.; Essex, D.W. Platelet-derived ERp57 mediates platelet incorporation into a growing thrombus by regulation of the alphaIIbbeta3 integrin. Blood 2013, 122, 3642–3650. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, Y.; Wang, L.; Rauova, L.; Hayes, V.M.; Poncz, M.; Essex, D.W. The disulfide isomerase ERp57 is required for fibrin deposition in vivo. J. Thromb. Haemost. 2014, 12, 1890–1897. [Google Scholar] [CrossRef]
- Munzer, P.; Walker-Allgaier, B.; Geue, S.; Geuss, E.; Hron, G.; Rath, D.; Eissler, D.; Winter, S.; Schaeffeler, E.; Meinert, M.; et al. PDK1 Determines Collagen-Dependent Platelet Ca2+ Signaling and Is Critical to Development of Ischemic Stroke In Vivo. Arter. Thromb. Vasc. Biol. 2016, 36, 1507–1516. [Google Scholar] [CrossRef]
- Manne, B.K.; Munzer, P.; Badolia, R.; Walker-Allgaier, B.; Campbell, R.A.; Middleton, E.; Weyrich, A.S.; Kunapuli, S.P.; Borst, O.; Rondina, M.T. PDK1 governs thromboxane generation and thrombosis in platelets by regulating activation of Raf1 in the MAPK pathway. J. Thromb. Haemost. 2018, 16, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; De, S.; Damron, D.S.; Chen, W.S.; Hay, N.; Byzova, V.T. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood 2004, 104, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Jurk, K.; Hobohm, L.; Jackel, S.; Saffarzadeh, M.; Schwierczek, K.; Wenzel, P.; Langer, F.; Reinhardt, C.; Ruf, W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 2017, 129, 2291–2302. [Google Scholar] [CrossRef] [PubMed]
- Senis, Y.A.; Tomlinson, M.G.; Ellison, S.; Mazharian, A.; Lim, J.; Zhao, Y.; Kornerup, K.N.; Auger, J.M.; Thomas, S.G.; Dhanjal, T.; et al. The tyrosine phosphatase CD148 is an essential positive regulator of platelet activation and thrombosis. Blood 2009, 113, 4942–4954. [Google Scholar] [CrossRef] [PubMed]
- Falet, H.; Pollitt, A.Y.; Begonja, A.J.; Weber, S.E.; Duerschmied, D.; Wagner, D.D.; Watson, S.P.; Hartwig, J.H. A novel interaction between FlnA and Syk regulates platelet ITAM-mediated receptor signaling and function. J. Exp. Med. 2010, 207, 1967–1979. [Google Scholar] [CrossRef]
- Adam, F.; Kauskot, A.; Nurden, P.; Sulpice, E.; Hoylaerts, M.F.; Davis, R.J.; Rosa, J.P.; Bryckaert, M. Platelet JNK1 is involved in secretion and thrombus formation. Blood 2010, 115, 4083–4092. [Google Scholar] [CrossRef]
- Pleines, I.; Hagedorn, I.; Gupta, S.; May, F.; Chakarova, L.; van Hengel, J.; Offermanns, S.; Krohne, G.; Kleinschnitz, C.; Brakebusch, C.; et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood 2012, 119, 1054–1063. [Google Scholar] [CrossRef]
- Xiang, B.; Zhang, G.; Ye, S.; Zhang, R.; Huang, C.; Liu, J.; Tao, M.; Ruan, C.; Smyth, S.S.; Whiteheart, S.W.; et al. Characterization of a Novel Integrin Binding Protein, VPS33B, Which Is Important for Platelet Activation and In Vivo Thrombosis and Hemostasis. Circulation 2015, 132, 2334–2344. [Google Scholar] [CrossRef]
- Sladojevic, N.; Oh, G.T.; Kim, H.H.; Beaulieu, L.M.; Falet, H.; Kaminski, K.; Freedman, J.E.; Liao, J.K. Decreased thromboembolic stroke but not atherosclerosis or vascular remodelling in mice with ROCK2-deficient platelets. Cardiovasc. Res. 2017, 113, 1307–1317. [Google Scholar] [CrossRef]
- Devanathan, V.; Hagedorn, I.; Kohler, D.; Pexa, K.; Cherpokova, D.; Kraft, P.; Singh, M.; Rosenberger, P.; Stoll, G.; Birnbaumer, L.; et al. Platelet Gi protein Galphai2 is an essential mediator of thrombo-inflammatory organ damage in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 6491–6496. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Yan, R.; Tian, J.; Chen, M.; Cui, Q.; Zhao, L.; Hu, R.; Jiang, M.; Li, Z.; et al. Receptor-interacting protein kinase 3 promotes platelet activation and thrombosis. Proc. Natl. Acad. Sci. USA 2017, 114, 2964–2969. [Google Scholar] [CrossRef] [PubMed]
- Yoda, E.; Rai, K.; Ogawa, M.; Takakura, Y.; Kuwata, H.; Suzuki, H.; Nakatani, Y.; Murakami, M.; Hara, S. Group VIB calcium-independent phospholipase A2 (iPLA2gamma) regulates platelet activation, hemostasis and thrombosis in mice. PLoS ONE 2014, 9, e109409. [Google Scholar] [CrossRef]
- Modjeski, K.L.; Ture, S.K.; Field, D.J.; Cameron, S.J.; Morrell, C.N. Glutamate Receptor Interacting Protein 1 Mediates Platelet Adhesion and Thrombus Formation. PLoS ONE 2016, 11, e0160638. [Google Scholar] [CrossRef]
- Taylor, K.A.; Wright, J.R.; Vial, C.; Evans, R.J.; Mahaut-Smith, M.P. Amplification of human platelet activation by surface pannexin-1 channels. J. Thromb. Haemost. 2014, 12, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Molica, F.; Meens, M.J.; Pelli, G.; Hautefort, A.; Emre, Y.; Imhof, B.A.; Fontana, P.; Scemes, E.; Morel, S.; Kwak, B.R. Selective inhibition of Panx1 channels decreases hemostasis and thrombosis in vivo. Thromb. Res. 2019, 183, 56–62. [Google Scholar] [CrossRef]
- Elaib, Z.; Adam, F.; Berrou, E.; Bordet, J.C.; Prevost, N.; Bobe, R.; Bryckaert, M.; Rosa, J.P. Full activation of mouse platelets requires ADP secretion regulated by SERCA3 ATPase-dependent calcium stores. Blood 2016, 128, 1129–1138. [Google Scholar] [CrossRef]
- Carubbi, C.; Mirandola, P.; Mattioli, M.; Galli, D.; Marziliano, N.; Merlini, P.A.; Lina, D.; Notarangelo, F.; Cozzi, M.R.; Gesi, M.; et al. Protein Kinase C ε Expression in Platelets from Patients with Acute Myocardial Infarction. PLOS ONE 2012, 7, e46409. [Google Scholar] [CrossRef]
- Unsworth, A.J.; Finney, B.A.; Navarro-Nunez, L.; Severin, S.; Watson, S.P.; Pears, C.J. Protein kinase Cepsilon and protein kinase Ctheta double-deficient mice have a bleeding diathesis. J. Thromb. Haemost. 2012, 10, 1887–1894. [Google Scholar] [CrossRef]
- Cardenas, E.I.; Gonzalez, R.; Breaux, K.; Da, Q.; Gutierrez, B.A.; Ramos, M.A.; Cardenas, R.A.; Burns, A.R.; Rumbaut, R.E.; Adachi, R. Munc18-2, but not Munc18-1 or Munc18-3, regulates platelet exocytosis, hemostasis, and thrombosis. J. Biol. Chem. 2019, 294, 4784–4792. [Google Scholar] [CrossRef]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef]
- Wei, S.; Wang, H.; Zhang, G.; Lu, Y.; An, X.; Ren, S.; Wang, Y.; Chen, Y.; White, J.G.; Zhang, C.; et al. Platelet IkappaB kinase-beta deficiency increases mouse arterial neointima formation via delayed glycoprotein Ibalpha shedding. Arter. Thromb. Vasc. Biol. 2013, 33, 241–248. [Google Scholar] [CrossRef]
- Hughan, S.C.; Spring, C.M.; Schoenwaelder, S.M.; Sturgeon, S.; Alwis, I.; Yuan, Y.; McFadyen, J.D.; Westein, E.; Goddard, D.; Ono, A.; et al. Dok-2 adaptor protein regulates the shear-dependent adhesive function of platelet integrin alphaIIbbeta3 in mice. J. Biol. Chem. 2014, 289, 5051–5060. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Vestweber, D.; Zarbock, A. GDF-15 prevents platelet integrin activation and thrombus formation. J. Thromb. Haemost. 2013, 11, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Liu, P.; Liu, Y.; Yue, M.; Wang, Y.; Wang, S.; Chen, X.; Zhou, Y.; Zhou, J.; Hu, X.; et al. Platelet Shp2 negatively regulates thrombus stability under high shear stress. J. Thromb. Haemost. 2019, 17, 220–231. [Google Scholar] [CrossRef]
- Cui, G.; Shan, L.; Guo, L.; Keung Chu, I.; Li, G.; Quan, Q.; Zhao, Y.; Meng Chong, C.; Zhang, Z.; Yu, P.; et al. Corrigendum: Novel anti-thrombotic agent for modulation of protein disulfide isomerase family member ERp57 for prophylactic therapy. Sci. Rep. 2015, 5, 13509. [Google Scholar] [CrossRef]
- Patel, P.; Naik, M.U.; Golla, K.; Shaik, N.F.; Naik, U.P. Calcium-induced dissociation of CIB1 from ASK1 regulates agonist-induced activation of the p38 MAPK pathway in platelets. Biochem. J. 2019, 476, 2835–2850. [Google Scholar] [CrossRef]
- Weng, Z.; Li, D.; Zhang, L.; Chen, J.; Ruan, C.; Chen, G.; Gartner, T.K.; Liu, J. PTEN regulates collagen-induced platelet activation. Blood 2010, 116, 2579–2581. [Google Scholar] [CrossRef]
- Patel, A.; Kostyak, J.; Dangelmaier, C.; Badolia, R.; Bhavanasi, D.; Aslan, J.E.; Merali, S.; Kim, S.; Eble, J.A.; Goldfinger, L.; et al. ELMO1 deficiency enhances platelet function. Blood Adv. 2019, 3, 575–587. [Google Scholar] [CrossRef]
- Wei, G.; Xu, X.; Tong, H.; Wang, X.; Chen, Y.; Ding, Y.; Zhang, S.; Ju, W.; Fu, C.; Li, Z.; et al. Salidroside inhibits platelet function and thrombus formation through AKT/GSK3beta signaling pathway. Aging 2020, 12, 8151–8166. [Google Scholar] [CrossRef]
- Hernandez, K.R.; Karim, Z.A.; Qasim, H.; Druey, K.M.; Alshbool, F.Z.; Khasawneh, F.T. Regulator of G-Protein Signaling 16 Is a Negative Modulator of Platelet Function and Thrombosis. J. Am. Heart Assoc. 2019, 8, e011273. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, G.; Xiang, B.; Chen, X.; Tang, L.; Shi, S.; Liu, Y.; Ai, X.; Xie, P.; Li, Z. TRAF3 negatively regulates platelet activation and thrombosis. Sci. Rep. 2017, 7, 17112. [Google Scholar] [CrossRef]
- Gupta, S.; Braun, A.; Morowski, M.; Premsler, T.; Bender, M.; Nagy, Z.; Sickmann, A.; Hermanns, H.M.; Bosl, M.; Nieswandt, B. CLP36 is a negative regulator of glycoprotein VI signaling in platelets. Circ. Res. 2012, 111, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, Y.; Zhou, J.; Chen, F.; Yang, A.; Essex, D.W. The transmembrane protein disulfide isomerase TMX1 negatively regulates platelet responses. Blood 2019, 133, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.L.; Wu, M.P.; Chung, C.L.; Yang, W.B.; Jayakumar, T.; Geraldine, P.; Chou, C.M.; Chang, C.Y.; Lu, W.J.; Sheu, J.R. Novel synthetic benzimidazole-derived oligosaccharide, M3BIM, prevents ex vivo platelet aggregation and in vivo thromboembolism. J. Biomed. Sci. 2016, 23, 26. [Google Scholar] [CrossRef]
- Chen, X.; Fan, X.; Tan, J.; Shi, P.; Wang, X.; Wang, J.; Kuang, Y.; Fei, J.; Liu, J.; Dang, S.; et al. Palladin is involved in platelet activation and arterial thrombosis. Thromb. Res. 2017, 149, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Wang, L.; Peng, R.; Zhao, Y.; Bai, F.; Yang, C.; Liu, X.; Wang, D.; Ma, B. Timosaponin AIII induces antiplatelet and antithrombotic activity via Gq-mediated signaling by the thromboxane A2 receptor. Sci. Rep. 2016, 6, 38757. [Google Scholar] [CrossRef]
- Kulkarni, P.P.; Tiwari, A.; Singh, N.; Gautam, D.; Sonkar, V.K.; Agarwal, V.; Dash, D. Aerobic glycolysis fuels platelet activation: Small-molecule modulators of platelet metabolism as anti-thrombotic agents. Haematologica 2019, 104, 806–818. [Google Scholar] [CrossRef]
- Janus-Bell, E.; Mangin, P.H. The relative importance of platelet integrins in hemostasis, thrombosis and beyond. Haematologica 2023, 108, 1734–1747. [Google Scholar] [CrossRef]
- Hagemeyer, C.E.; Peter, K. Targeting the platelet integrin GPIIb/IIIa. Curr. Pharm. Des. 2010, 16, 4119–4133. [Google Scholar] [CrossRef]
- Schwarz, M.; Meade, G.; Stoll, P.; Ylanne, J.; Bassler, N.; Chen, Y.C.; Hagemeyer, C.E.; Ahrens, I.; Moran, N.; Kenny, D.; et al. Conformation-specific blockade of the integrin GPIIb/IIIa: A novel antiplatelet strategy that selectively targets activated platelets. Circ. Res. 2006, 99, 25–33. [Google Scholar] [CrossRef]
- Fagerholm, S.C.; Lek, H.S.; Morrison, V.L. Kindlin-3 in the immune system. Am. J. Clin. Exp. Immunol. 2014, 3, 37–42. [Google Scholar] [PubMed]
- Rezaie, Y.; Fattahi, F.; Mashinchi, B.; Kamyab Hesari, K.; Montazeri, S.; Kalantari, E.; Madjd, Z.; Saeednejad Zanjani, L. High expression of Talin-1 is associated with tumor progression and recurrence in melanoma skin cancer patients. BMC Cancer 2023, 23, 302. [Google Scholar] [CrossRef]
- Wu, Y.; Ahmad, S.S.; Zhou, J.; Wang, L.; Cully, M.P.; Essex, D.W. The Disulfide Isomerase Erp57 Mediates Platelet Aggregation and Hemostasis. Blood 2011, 118, 1129. [Google Scholar] [CrossRef]
- Reddy, K.B.; Smith, D.M.; Plow, E.F. Analysis of fyn function in hemostasis and αllbβ3-integrin signaling. J. Cell Sci. 2008, 121, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M.; Smyth, S.S.; Schoenwaelder, S.M.; Fischer, T.H.; White 2nd, G.C. Rap1b is required for normal platelet function and hemostasis in mice. J. Clin. Investig. 2005, 115, 680–687. [Google Scholar] [CrossRef]
- Stritt, S.; Wolf, K.; Lorenz, V.; Vögtle, T.; Gupta, S.; Bösl, M.R.; Nieswandt, B. Rap1-GTP-interacting adaptor molecule (RIAM) is dispensable for platelet integrin activation and function in mice. Blood 2015, 125, 219–222. [Google Scholar] [CrossRef]
- Aslan, J.E.; Mccarty, O.J.T. Rho GTPases in platelet function. J. Thromb. Haemost. 2012, 11, 35–46. [Google Scholar] [CrossRef]
- Wang, L.; Wu, J.; Zhang, W.; Zhi, Y.; Wu, Y.; Jiang, R.; Yang, R. Effects of aspirin on the ERK and PI3K/Akt signaling pathways in rats with acute pulmonary embolism. Mol. Med. Rep. 2013, 8, 1465–1471. [Google Scholar]
- Rathore, V.; Stapleton, M.A.; Hillery, C.A.; Montgomery, R.R.; Nichols, T.C.; Merricks, E.P.; Newman, D.K.; Newman, P.J. PECAM-1 negatively regulates GPIb/V/IX signaling in murine platelets. Blood 2003, 102, 3658–3664. [Google Scholar] [CrossRef]
- Li, D.; August, S.; Woulfe, D.S. GSK3beta is a negative regulator of platelet function and thrombosis. Blood 2008, 111, 3522–3530. [Google Scholar]
- Goschnick, M.W.; Lau, L.M.; Wee, J.L.; Liu, Y.S.; Hogarth, P.M.; Robb, L.M.; Hickey, M.J.; Wright, M.D.; Jackson, D.E. Impaired “outside-in” integrin alphaIIbbeta3 signaling and thrombus stability in TSSC6-deficient mice. Blood 2006, 108, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Flevaris, P.; Stojanovic, A.; Gong, H.; Chishti, A.; Welch, E.; Du, X. A molecular switch that controls cell spreading and retraction. J. Cell Biol. 2007, 179, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Leitges, M.; Stegner, D. Protein kinase Ciota/lambda is dispensable for platelet function in thrombosis and hemostasis in mice. Cell Signal. 2017, 38, 223–229. [Google Scholar] [PubMed]
- Farese, R.V.; Sajan, M.P.; Yang, H.; Li, P.; Mastorides, S.; Gower, W.R., Jr.; Nimal, S.; Choi, C.S.; Kim, S.; Shulman, G.I.; et al. Muscle-specific knockout of PKC-λ impairs glucose transport and induces metabolic and diabetic syndromes. J. Clin. Investig. 2007, 117, 3141. [Google Scholar] [CrossRef]
- Salzmann, M.; Bleichert, S.; Moser, B.; Mussbacher, M.; Haase, M.; Hoesel, B.; Schrottmaier, W.C.; Kral-Pointner, J.B.; Itakura, M.; Schmidt, K.; et al. IkappaB kinase 2 is not essential for platelet activation. Blood Adv. 2020, 4, 638–643. [Google Scholar] [CrossRef]
- Fischer, K.C.; Daunt, C.P.; Tremblay, C.S.; Dias, S.; Vince, J.E.; Jabbour, A.M. Deletion of IKK2 in haematopoietic cells of adult mice leads to elevated interleukin-6, neutrophilia and fatal gastrointestinal inflammation. Cell Death Dis. 2021, 12, 28. [Google Scholar] [CrossRef]
- Schurr, Y.; Spindler, M.; Kurz, H.; Bender, M. The cytoskeletal crosslinking protein MACF1 is dispensable for thrombus formation and hemostasis. Sci. Rep. 2019, 9, 7726. [Google Scholar] [CrossRef]
- Kang, L.; Liu, Y.; Jin, Y.; Li, M.; Song, J.; Zhang, Y.; Zhang, Y.; Yang, Y. Mutations of MACF1, Encoding Microtubule-Actin Crosslinking-Factor 1, Cause Spectraplakinopathy. Front. Neurol. 2019, 10, 1335. [Google Scholar] [CrossRef]
- Uemura, T.; Kawasaki, T.; Taniguchi, M.; Moritani, Y.; Hayashi, K.; Saito, T.; Takasaki, J.; Uchida, W.; Miyata, K. Biological properties of a specific Galpha q/11 inhibitor, YM-254890, on platelet functions and thrombus formation under high-shear stress. Br. J. Pharmacol. 2006, 148, 61–69. [Google Scholar] [CrossRef]
- Salles Ii, C.; Monkman, J.H.; Ahnstrom, J.; Lane, D.A.; Crawley, J.T. Vessel wall BAMBI contributes to hemostasis and thrombus stability. Blood 2014, 123, 2873–2881. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, C.; Xu, Y.; Huang, K.; Wang, Y.; Wang, X.; Zhou, X.; Pang, W.; Yang, G.; Yu, T. Adipose-specific BMP and activin membrane-bound inhibitor (BAMBI) deletion promotes adipogenesis by accelerating ROS production. J. Biol. Chem. 2021, 296, 100037. [Google Scholar] [CrossRef]
- Bhatia, K.; Jain, V.; Aggarwal, D.; Vaduganathan, M.; Arora, S.; Hussain, Z.; Uberoi, G.; Tafur, A.; Zhang, C.; Ricciardi, M.; et al. Dual Antiplatelet Therapy Versus Aspirin in Patients With Stroke or Transient Ischemic Attack: Meta-Analysis of Randomized Controlled Trials. Stroke 2021, 52, e217–e223. [Google Scholar] [CrossRef] [PubMed]
- Agbaedeng, T.A.; Noubiap, J.J.; Roberts, K.A.; Chew, D.P.; Psaltis, P.J.; Amare, A.T. Sex-Based Outcomes of Dual-Antiplatelet Therapy After Percutaneous Coronary Intervention: A Pairwise and Network Meta-Analysis. Drugs 2024, 84, 685–701. [Google Scholar] [CrossRef]
- Liu, J.; Lu, Y.; Zheng, B.; Huang, D.; Song, J.; Wang, B.; Zheng, S. Talin1 promotes HCC progression by regulating NRG1/PI3K/AKT pathway. Discov. Oncol. 2024, 15, 360. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Padovani, A.; Borroni, B.; Colciaghi, F.; Pastorino, L.; Archetti, S.; Cottini, E.; Caimi, L.; Cattabeni, F.; Di Luca, M. Platelet amyloid precursor protein forms in AD: A peripheral diagnostic tool and a pharmacological target. Mech. Ageing Dev. 2001, 122, 1997–2004. [Google Scholar] [CrossRef]
- Berger, J.S.; Cornwell, M.G.; Xia, Y.; Muller, M.A.; Smilowitz, N.R.; Newman, J.D.; Schlamp, F.; Rockman, C.B.; Ruggles, K.V.; Voora, D.; et al. A Platelet Reactivity ExpreSsion Score derived from patients with peripheral artery disease predicts cardiovascular risk. Nat. Commun. 2024, 15, 6902. [Google Scholar] [CrossRef]
- Pereira, N.L.; Cresci, S.; Angiolillo, D.J.; Batchelor, W.; Capers, Q.t.; Cavallari, L.H.; Leifer, D.; Luzum, J.A.; Roden, D.M.; Stellos, K.; et al. CYP2C19 Genetic Testing for Oral P2Y12 Inhibitor Therapy: A Scientific Statement From the American Heart Association. Circulation 2024, 150, e129–e150. [Google Scholar] [CrossRef]
- Schattner, M.; Rabinovich, G.A. Galectins: New agonists of platelet activation. Biol. Chem. 2013, 394, 857–863. [Google Scholar] [CrossRef]
- Zapilko, V.; Fish, R.J.; Garcia, A.; Reny, J.-L.; Dunoyer-Geindre, S.; Lecompte, T.; Neerman-Arbez, M.; Fontana, P. MicroRNA-126 is a regulator of platelet-supported thrombin generation. Platelets 2020, 31, 746–755. [Google Scholar] [CrossRef]
- Sepúlveda, M.; Palomo, I.; Montecino-Garrido, H.; Wehinger, S.; Rodriguez-Mañas, L.; Trostchansky, A.; Fuentes, E. Physiological changes associated with aging: Identification of novel biomarkers for frailty syndrome in women. Free Radic. Biol. Med. 2024, 223, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Burla, B.; Oh, J.; Nowak, A.; Piraud, N.; Meyer, E.; Mei, D.; Bendt, A.K.; Studt, J.D.; Frey, B.M.; Torta, F.; et al. Plasma and platelet lipidome changes in Fabry disease. Clin. Chim. Acta Int. J. Clin. Chem. 2024, 562, 119833. [Google Scholar] [CrossRef]
- Xiao, Z.; Lin, M.; Song, N.; Wu, X.; Hou, J.; Wang, L.; Tian, X.; An, C.; Dela Cruz, C.S.; Sharma, L.; et al. Clinical features and multiomics profiles indicate coagulation and platelet dysfunction in COVID-19 viral sepsis. iScience 2024, 27, 110110. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Shen, H.; Cao, F.; He, H.; Li, B.; Zhang, H.; Zhang, X.; Li, Z. Bioinformatics Analysis Reveals Crosstalk Among Platelets, Immune Cells, and the Glomerulus That May Play an Important Role in the Development of Diabetic Nephropathy. Front. Med. 2021, 8, 657918. [Google Scholar] [CrossRef]
- Go, S.; Jeong, D.; Chung, J.; Kim, G.-h.; Song, J.; Moon, E.; Huh, Y.H.; Kim, D. Super-resolution imaging reveals cytoskeleton-dependent organelle rearrangement within platelets at intermediate stages of maturation. Structure 2021, 29, 810–822.e3. [Google Scholar] [CrossRef]
- Chung, J.; Jeong, D.; Kim, G.-H.; Go, S.; Song, J.; Moon, E.; Huh, Y.H.; Kim, D. Super-resolution imaging of platelet-activation process and its quantitative analysis. Sci. Rep. 2021, 11, 10511. [Google Scholar] [CrossRef]
- Junior, A.S.F.; Lessa, M.P.M.; Sanborn, K.; Gordee, A.; Kuchibhatla, M.; Karafin, M.S.; Onwuemene, O.A. Bleeding recurrence risk among hospitalized patients undergoing therapeutic plasma exchange: A multi-center study. Blood Transfus. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Liu, L.; Liu, D.; He, T.; Liang, B.; Zhao, J. Coagulation risk predicting in anticoagulant-free CRRT. Blood Purif. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Grover, S.P.; Hisada, Y.M.; Kasthuri, R.S.; Reeves, B.N.; Mackman, N. Cancer Therapy–Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1291–1305. [Google Scholar] [CrossRef]
- Haddad, T.C.; Greeno, E.W. Chemotherapy-induced thrombosis. Thromb. Res. 2006, 118, 555–568. [Google Scholar] [CrossRef]
- Middleton, E.A.; Weyrich, A.S.; Zimmerman, G.A. Platelets in Pulmonary Immune Responses and Inflammatory Lung Diseases. Physiol. Rev. 2016, 96, 1211–1259. [Google Scholar] [CrossRef] [PubMed]
- Karolczak, K.; Soltysik, B.; Kostka, T.; Witas, P.J.; Watala, C. Platelet and Red Blood Cell Counts, as well as the Concentrations of Uric Acid, but Not Homocysteinaemia or Oxidative Stress, Contribute Mostly to Platelet Reactivity in Older Adults. Oxidative Med. Cell. Longev. 2019, 2019, 9467562. [Google Scholar] [CrossRef]
- Przygodzki, T.; Luzak, B.; Kassassir, H.; Mnich, E.; Boncler, M.; Siewiera, K.; Kosmalski, M.; Szymanski, J.; Watala, C. Diabetes and Hyperglycemia Affect Platelet GPIIIa Expression. Effects on Adhesion Potential of Blood Platelets from Diabetic Patients under In Vitro Flow Conditions. Int. J. Mol. Sci. 2020, 21, 3222. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.L.; Dunster, J.L.; Kriek, N.; Unsworth, A.J.; Sage, T.; Mohammed, Y.M.M.; De Simone, I.; Taylor, K.A.; Bye, A.P.; Ólafsson, G.; et al. The rate of platelet activation determines thrombus size and structure at arterial shear. J. Thromb. Haemost. 2023, 21, 2248–2259. [Google Scholar] [CrossRef] [PubMed]
- Gilanchi, S.; Faranoush, M.; Daskareh, M.; Sadjjadi, F.S.; Zali, H.; Ghassempour, A.; Rezaei Tavirani, M. Proteomic-Based Discovery of Predictive Biomarkers for Drug Therapy Response and Personalized Medicine in Chronic Immune Thrombocytopenia. BioMed Res. Int. 2023, 2023, 9573863. [Google Scholar] [CrossRef]
- Grande, R.; Dovizio, M.; Marcone, S.; Szklanna, P.B.; Bruno, A.; Ebhardt, H.A.; Cassidy, H.; F, N.Á.; Caprodossi, A.; Lanuti, P.; et al. Platelet-Derived Microparticles From Obese Individuals: Characterization of Number, Size, Proteomics, and Crosstalk With Cancer and Endothelial Cells. Front. Pharmacol. 2019, 10, 7. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Eguchi, A.; Imami, K.; Tempaku, M.; Izuoka, K.; Takase, T.; Kainuma, K.; Nagao, M.; Furuta, N.; Iwasa, M.; et al. Circulating extracellular vesicles are associated with pathophysiological condition including metabolic syndrome-related dysmetabolism in children and adolescents with obesity. J. Mol. Med. 2023, 102, 23–38. [Google Scholar] [CrossRef]
- El-Sayed, H.A.; Othman, M.; Azzam, H.; Bucciol, R.; Ebrahim, M.A.; El-Agdar, M.; Tera, Y.; Sakr, D.H.; Ghoneim, H.R.; Selim, T.E. Assessing the risk of venous thromboembolism in patients with haematological cancers using three prediction models. J. Cancer Res. Clin. Oncol. 2023, 149, 17771–17780. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Pathway | Role | Diseases | References |
|---|---|---|---|
| Granule secretion | This pathway contemplates proteins used for cytoskeletal organization, intracellular calcium levels, kinase activity, and intracellular protease activity, which facilitate membrane fusion and granule secretion from platelets. | Stroke, cerebral infarction, chronic inflammatory bowel disease, sepsis, lupus, atherosclerosis, acute malaria, T-cell dysfunction in HIV-1. | [14,15,16,17] |
| Integrin | Integrins are receptors in the membrane that mediate signals in response to the extracellular matrix, such as motility, cell shape, and cell cycle progression. | Thrombosis, coronary artery disease, inflammatory bowel disease, glioblastoma, dry eye disease, cancer, fibrosis, respiratory and neurological diseases, multiple sclerosis, SARS-Cov-2 infection, diabetic retinopathy, glaucoma, optic nerve degeneration, Alzheimer’s disease, Crohn’s disease, nephropathy, and asthma. | [18,19,20,21,22,23,24,25,26,27,28,29,30] |
| GPCRs pathway | GPCR binds external molecules, activating G proteins and activating or inhibiting pathways for CaMP signaling and phosphatidylinositol signaling. | Retinitis pigmentosa, nephrogenic diabetes insipidus, obesity, heart failure, asthma, hypo- and hyperthyroidism, fertility disorders, and carcinoma. | [31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46] |
| MAPKs pathway | The pathway of MAPK/ERK transduces signals downstream of cytokine receptors and regulates the transcription of specific proteins and the integrin outside-in pathway. | Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, cancer, and lymphoma. | [47,48,49,50,51,52,53,54,55,56,57,58,59,60] |
| Rho/ROCK pathway | The ROCK pathway involves the use of serine–threonine protein kinase in cell movement and reorganization of the cytoskeleton. | Alzheimer’s disease, Parkinson’s disease, ischemic myocardial fibrosis, atherosclerosis, restenosis, hypertension, pulmonary hypertension, cardiac hypertrophy, amyotrophic lateral sclerosis, and cancer. | [61,62,63,64,65,66,67,68,69,70,71] |
| Tyrosine kinase pathways | Tyrosine kinases are cell-surface proteins that act as signal transducers, regulating cellular processes such as apoptosis, proliferation, metabolism, and differentiation. | Cancer, vascular diseases, fibrosis, immune-mediated disorders, rheumatoid arthritis, bone disorders, arteriosclerosis, Alzheimer’s disease, and Parkinson’s disease. | [72,73,74,75,76,77,78,79] |
| Thromboxane pathway | Thromboxane synthesis induces pro-inflammatory and pro-thrombotic phenotypes by metabolizing PUFA from the membrane. | Atherosclerosis, hypertension, diabetes, obesity, myocardial infarction, and stroke. | [80,81,82,83,84,85] |
| Protein | Role | Type of Protein | Pathway Involved | Target | Depletion Experiment | Reference |
|---|---|---|---|---|---|---|
| Integrin αIIβ1 | Activatory | Integrin | Integrin αIIβ1 | αIIβ1-/- | [97] | |
| Integrin αIIbβ3 | Activatory | Integrin | Integrin αIIbβ3 | αIIbβ3-/- | [97] | |
| Kindlin-3 | Activatory | Signaling protein | Integrin αIIbβ3 | Talin | KIND3-/- | [97] |
| Dok-1 | Activatory | adapter protein | Integrin αIIbβ3 | integrin αIIbβ3 | Dok-1-/- | [98] |
| Talin-1 | Activatory | Cytoskeletal protein | Integrin αIIbβ3 | integrin β1 and β3 | Talin-1 (L325R) | [99,100,101] |
| Rap1 | Activatory | Gtpase | Integrin αIIbβ3 | Talin-1 | Rap1-/- | [102] |
| ILK | Activatory | Kinase | Integrin αIIbβ3 | integrin β1 and β3 | ILK-/- | [103,104] |
| CD154 | Activatory | Ligand of CD40 | Integrin αIIbβ3 | CD40 | CD154-/- | [105] |
| ASK1 | Activatory | MAP kinase | Integrin αIIbβ3 | p38 MAPK | ASK1-/- | [106,107] |
| ERp57 | Activatory | Disulfide isomerase | Integrin αIIbβ3 | integrin αIIbβ3 | ERp57-/- anti-ERp57 | [108,109,110,111] |
| PDK1 | Activatory | Kinase | P2Y12 | raf1 | PDK1-/- | [112,113] |
| AKT1 | Activatory | Serine/threonine-protein kinase | AKT/GSK3β | GSK3β | AKT1-/- | [114] |
| Factor 3 (C3) | Activatory | Cofactor | Complement | Rap1b | C3-/- | [115] |
| Fyn | Activatory | Src family | Integrin αIIbβ3 | Panx1 | Fyn-/- | |
| CD148 | Activatory | Protein Tyrosine Phosphatase | GPVI and SFK | GPVI | CD148-/- | [116] |
| Filamin | Activatory | Actin-binding protein | Spreading and granule release | Actin | FlnA-/- | [117] |
| JNK1 | Activatory | MAP kinase | MEK/ERK | PKC | JNK1-/- | [118] |
| RhoA | Activatory | GTPase | RhoA-ROCK-MLC | ROCK | RhoA-/- | [119] |
| VPS33B | Activatory | Sorting protein | RhoA-ROCK-MLC | integrin | VPS33B-/- | [120] |
| ROCK | Activatory | Serine/threonine kinase | RhoA-ROCK-MLC | Actin | ROCK2Plt-/- | [121] |
| Gα(i2) | Activatory | G protein | GPCRs | Adenylate cyclase | Gα(i2)-/- | [122] |
| Gα13 | Activatory | G protein | GPCRs | RhoA | Gα13-/- | |
| RIP3 | Activatory | serine/threonine kinase | GPCRs | Gα13 | RIP3 inhibitor | [123] |
| Rap1b | Activatory | GTPase | GPCRs | RIAMS preading | Rap1b-/- | |
| iPLA2γ | Activatory | Phospholipase | TXA2 production | Fatty acids | iPLA2γ-/- | [124] |
| GRIP1 | Activatory | Receptor interacting protein | GPIb-IX, GPIbα, GPIbβ, GPIb-IX | GPIb-IX, GPIbα, GPIbβ, GPIb-IX | GRIP1-/- | [125] |
| TSSC6 | Activatory | Integral membrane protein | Integrin | integrin αIIbβ3 | TSSC6-/- | |
| Panx1 | Activatory | ATP channel | integrin αIIbβ3 | P2X1 | Panx-/- PanxPDEL BrilliantBlue-FCF | [126,127] |
| SERCA3 | Activatory | Intracelular Ca2+ pump ATPase | Granule secretion | Ca2+ | SERCA3-/- | [128] |
| PKC | Activatory | Kinase | GPVI and MEK/ERK | PAR1, ERK | PKCθ-/- and PKCε-/- | [129,130] |
| Munc18-2 | Activatory | Vesicle docking protein | Granule secretion | SNARE | Munc18-2-/- | [131] |
| HMGB1 | Activatory | DNA binding protein | TLR4/cGKI | TLR4/cGKI | HMGB1-/- | [132] |
| IKKβ | Activatory | IκB kinase | Integrin αIIbβ3 | IκB | IKKβ-/- | [133] |
| Dok-2 | Negative | Adapter protein | Tyrosine kinase | integrin αIIbβ3 | Dok-2-/- | [134] |
| PP2B | Negative | Phosphatase | Src | Filamin A | PP2B-Aβ-/- PP2B-Aβ depleted | [93] |
| GDF-15 | Negative | Growth factor | integrin αIIbβ3 and αIIβ1 | PKA | GDF-15-/- | [135] |
| Shp2 | Negative | Tyrosine phosphatase | integrin αIIbβ3 and αIIβ1 | AKT | Shp2-/- | [136] |
| ADTM | Inhibitory | Compound | integrin αIIbβ3 | ERp57 | [137] | |
| CIB1 | Negative | Calcium and integrin binding | integrin αIIbβ3 | ASK1 | CIB1-/- | [138] |
| PTEN | Negative | Phosphatase | AKT/GSK3β | PI3K | PTEN-/- | [139] |
| ELMO1 | Negative | RAC1 activator | RhoG | RAC1 | ELMO1-/- | [140] |
| PECAM-1 | Negative | Transmembrane glycoprotein | GPIb/V/IX integrin AKT/GSK3β | PI3K | PECAM-1-/- | |
| GSK3β | Negative | Kinase | AKT/GSK3β | b Catenin | GSK3β-/- | |
| Salidroside | Inhibitory | Compound | AKT/GSK3β | Mitochondria | [141] | |
| RGS 16 | Negative | G protein regulator | GPCRs | Gi | RGS-16-/- | [142] |
| TRAF3 | Negative | TNFR-associated factor | Integrin αIIbβ3 | CD40 | TRAF3-/- | [143] |
| CLP36 | Negative | Cytoskeleton-associated protein | GPVI | GPVI | CLP36-/- CLP36 (ΔLIM) | [144] |
| TMX1 | Negative | Transmembrane protein | integrin αIIbβ3 | integrin αIIbβ3 | TMX1-/- | [145] |
| M3BIM | Inhibitory | Compound | PLC/PKC/MAPKs | PKC | [146] | |
| palladin | Negative | Actin-associated protein | Spreading | RAC1 | palladin-/+ | [147] |
| TAIII | Inhibitory | Compound | TP receptor | PKC | [148] | |
| DCa | Inhibitory | Compound | AKT/GSK3β | Mitochondria | [149] | |
| DHEA | Inhibitory | Compound | AKT/GSK3β | Mitochondria | [149] | |
| DASA | Inhibitory | Compound | AKT/GSK3β | Mitochondria | [149] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montecino-Garrido, H.; Trostchansky, A.; Espinosa-Parrilla, Y.; Palomo, I.; Fuentes, E. How Protein Depletion Balances Thrombosis and Bleeding Risk in the Context of Platelet’s Activatory and Negative Signaling. Int. J. Mol. Sci. 2024, 25, 10000. https://doi.org/10.3390/ijms251810000
Montecino-Garrido H, Trostchansky A, Espinosa-Parrilla Y, Palomo I, Fuentes E. How Protein Depletion Balances Thrombosis and Bleeding Risk in the Context of Platelet’s Activatory and Negative Signaling. International Journal of Molecular Sciences. 2024; 25(18):10000. https://doi.org/10.3390/ijms251810000
Chicago/Turabian StyleMontecino-Garrido, Hector, Andrés Trostchansky, Yolanda Espinosa-Parrilla, Iván Palomo, and Eduardo Fuentes. 2024. "How Protein Depletion Balances Thrombosis and Bleeding Risk in the Context of Platelet’s Activatory and Negative Signaling" International Journal of Molecular Sciences 25, no. 18: 10000. https://doi.org/10.3390/ijms251810000