

Tracking Response and Resistance in Acute Myeloid Leukemia through Single-Cell DNA Sequencing Helps Uncover New Therapeutic Targets

 , , , ,
, , , ,  ,
,  ,
,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Results
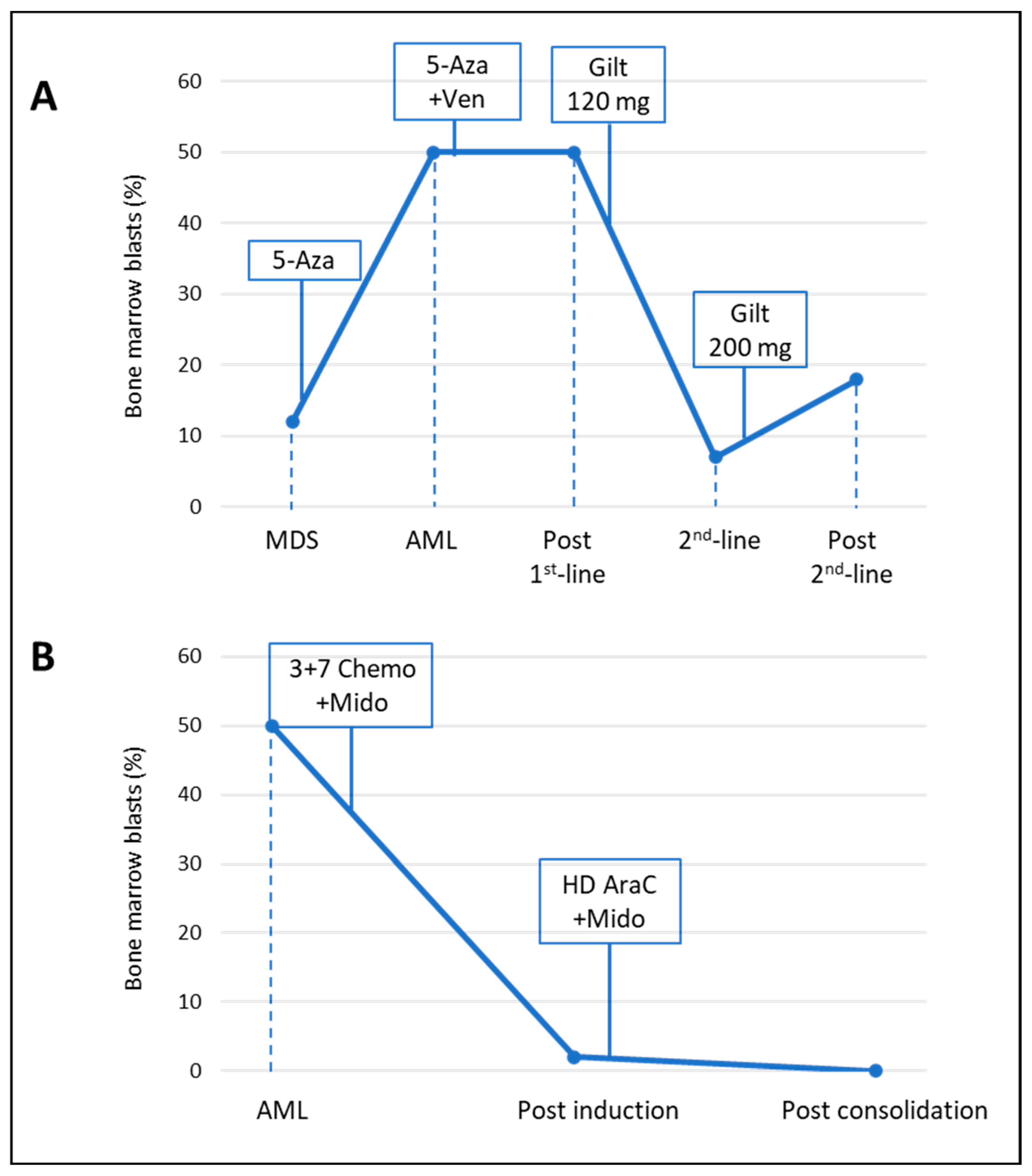
2.1. Clinical, Cytogenetic and Molecular Patients’ History
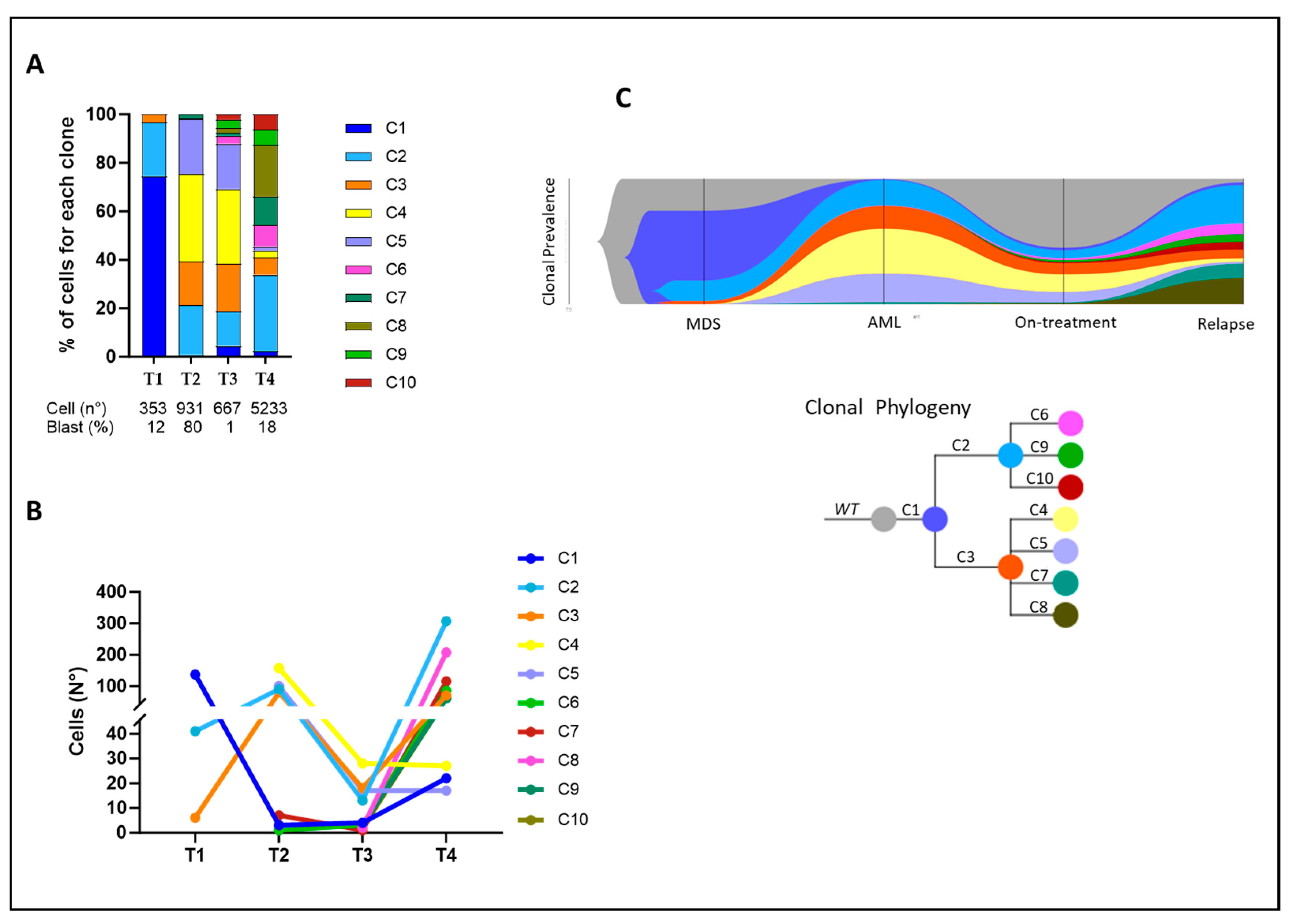
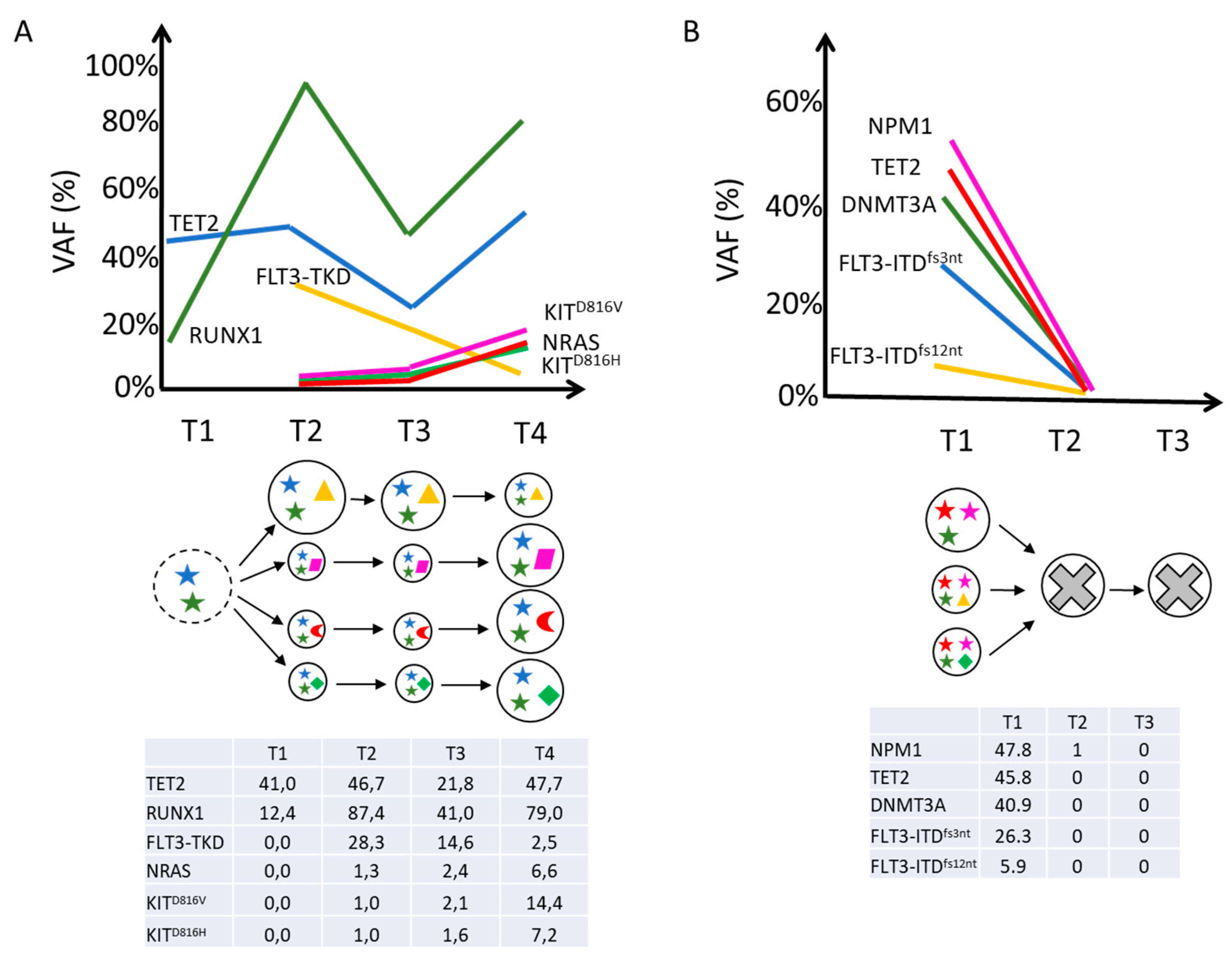
2.2. Clonal Architecture and Evolution of FLT3-Mutated AML
3. Discussion
4. Materials and Methods
4.1. Patients, Samples, and Cell Preparation
4.2. Bulk Next-Generation DNA Sequencing (NGS)
4.3. Single-Cell DNA Sequencing (scDNAseq)
4.4. Data Processing and Variant Filtering
4.5. Variant Calling
4.6. Clonal Architecture Analysis
4.7. Copy Number Analysis (CNA)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Rosnet, O.; Bühring, H.J.; Marchetto, S.; Rappold, I.; Lavagna, C.; Sainty, D.; Arnoulet, C.; Chabannon, C.; Kanz, L.; Hannum, C.; et al. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 1996, 10, 238–248. [Google Scholar] [PubMed]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef]
- Tsapogas, P.; Mooney, C.J.; Brown, G.; Rolink, A. The Cytokine Flt3-Ligand in Normal and Malignant Hematopoiesis. Int. J. Mol. Sci. 2017, 18, 1115. [Google Scholar] [CrossRef]
- Beaudin, A.E.; Boyer, S.W.; Forsberg, E.C. Flk2/Flt3 promotes both myeloid and lymphoid development by expanding non–self-renewing multipotent hematopoietic progenitor cells. Exp. Hematol. 2013, 42, 218–229.e4. [Google Scholar] [CrossRef]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef]
- German-Austrian AML Study Group (AMLSG); Nagel, G.; Weber, D.; Fromm, E.; Erhardt, S.; Lübbert, M.; Fiedler, W.; Kindler, T.; Krauter, J.; Brossart, P.; et al. Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO). Ann. Hematol. 2017, 96, 1993–2003. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Ambinder, A.J.; Levis, M. Potential targeting of FLT3 acute myeloid leukemia. Haematologica 2020, 106, 671–681. [Google Scholar] [CrossRef]
- Friedman, R. The molecular mechanisms behind activation of FLT3 in acute myeloid leukemia and resistance to therapy by selective inhibitors. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1877, 188666. [Google Scholar] [CrossRef]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Voso, M.T.; Larson, R.A.; Jones, D.; Marcucci, G.; Prior, T.; Krauter, J.; Heuser, M.; Lavorgna, S.; Nomdedeu, J.; Geyer, S.M.; et al. Midostaurin in patients with acute myeloid leukemia and FLT3-TKD mutations: A subanalysis from the RATIFY trial. Blood Adv. 2020, 4, 4945–4954. [Google Scholar] [CrossRef] [PubMed]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2019, 111, 312–322. [Google Scholar] [CrossRef]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.; De, O.; Berti, E.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Juliusson, G.; Jädersten, M.; Deneberg, S.; Lehmann, S.; Möllgård, L.; Wennström, L.; Antunovic, P.; Cammenga, J.; Lorenz, F.; Ölander, E.; et al. The prognostic impact of FLT3-ITD and NPM1 mutation in adult AML is age-dependent in the population-based setting. Blood Adv. 2020, 4, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Negotei, C.; Colita, A.; Mitu, I.; Lupu, A.R.; Lapadat, M.-E.; Popovici, C.E.; Crainicu, M.; Stanca, O.; Berbec, N.M. A Review of FLT3 Kinase Inhibitors in AML. J. Clin. Med. 2023, 12, 6429. [Google Scholar] [CrossRef]
- Kennedy, V.E.; Smith, C.C. FLT3 targeting in the modern era: From clonal selection to combination therapies. Int. J. Hematol. 2023, 1–13. [Google Scholar] [CrossRef]
- Short, N.J.; Nguyen, D.; Ravandi, F. Treatment of older adults with FLT3-mutated AML: Emerging paradigms and the role of frontline FLT3 inhibitors. Blood Cancer J. 2023, 13, 1–9. [Google Scholar] [CrossRef]
- Capelli, D. FLT3-Mutated Leukemic Stem Cells: Mechanisms of Resistance and New Therapeutic Targets. Cancers 2024, 16, 1819. [Google Scholar] [CrossRef]
- Capelli, D.; Menotti, D.; Fiorentini, A.; Saraceni, F.; Olivieri, A. Overcoming Resistance: FLT3 Inhibitors Past, Present, Future and the Challenge of Cure. Cancers 2022, 14, 4315. [Google Scholar] [CrossRef]
- Fedorov, K.; Maiti, A.; Konopleva, M. Targeting FLT3 Mutation in Acute Myeloid Leukemia: Current Strategies and Future Directions. Cancers 2023, 15, 2312. [Google Scholar] [CrossRef]
- Borsi, E.; Vigliotta, I.; Poletti, A.; Mazzocchetti, G.; Solli, V.; Zazzeroni, L.; Martello, M.; Armuzzi, S.; Taurisano, B.; Kanapari, A.; et al. Single-Cell DNA Sequencing Reveals an Evolutionary Pattern of CHIP in Transplant Eligible Multiple Myeloma Patients. Cells 2024, 13, 657. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt 1 | Pt 2 | |
|---|---|---|
| Gender | Male | Female |
| Age at diagnosis (y) | 82 | 50 |
| AML type | Secondary to MDS | De novo |
| WBC at diagnosis (103 × µL) | 111.370 | 30.000 |
| Peripheral blasts (%) | 69 | 45 |
| Bone marrow blasts (%) | 50 | 50 |
| ELN Risk category | Adverse | Intermediate |
| FLT3 mutational status | TKD positive (D835H) | ITD positive |
| Additional mutations | ASXL1, SRSF2, TET2, RUNX1 | NPM1 |
| Cytogenetics | NK (46, XY) | NK (46, XX) |
| First-line therapy | azacitidine plus venetoclax | Intensive chemotherapy plus midostaurin |
| Second line therapy | gilteritinib | N.A. |
| Consolidation therapy | N.A. | High-dose cytarabine plus midostaurin |
| Clone | Pathogenic Variants | Time Point | Proportion (%) |
|---|---|---|---|
| C1 | TET2N191Kfs*4 | T1 | 38.1 |
| T2 | 0.32 | ||
| T3 | 0.6 | ||
| T4 | 0.42 | ||
| C2 | TET2N191Kfs*4 RUNX1R201Q_HET | T1 | 11.61 |
| T2 | 9.67 | ||
| T3 | 1.94 | ||
| T4 | 5.87 | ||
| C3 | TET2N191Kfs*4 RUNX1R201Q _HOM | T1 | 1.7 |
| T2 | 8.59 | ||
| T3 | 2.68 | ||
| T4 | 1.34 | ||
| C4 | TET2N191Kfs*4; RUNX1R201Q _HOM; FLT3D835H | T1 | 0 |
| T2 | 16.86 | ||
| T3 | 4.17 | ||
| T4 | 0.52 | ||
| C5 | TET2N191Kfs*4 RUNX1R201Q_HET; FLT3D835H | T1 | 0 |
| T2 | 10.74 | ||
| T3 | 2.53 | ||
| T4 | 0.32 | ||
| C6 | TET2N191Kfs*4 RUNX1R201Q _HOM; NRASG12A | T1 | 0 |
| T2 | 0.11 | ||
| T3 | 0.45 | ||
| T4 | 1.62 | ||
| C7 | TET2N191Kfs*4 RUNX1R201Q_HOM; KITD816H | T1 | 0 |
| T2 | 0.75 | ||
| T3 | 0.15 | ||
| T4 | 2.2 | ||
| C8 | TET2N191Kfs*4 RUNX1R201Q _HOM; KITD816V | T1 | 0 |
| T2 | 0 | ||
| T3 | 0.3 | ||
| T4 | 3.96 | ||
| C9 | TET2N191Kfs*4 RUNX1R201Q_HET; KITD816H | T1 | 0 |
| T2 | 0 | ||
| T3 | 0.45 | ||
| T4 | 1.17 | ||
| C10 | TET2N191Kfs*4 RUNX1R201Q_HET; NRASG12A | T1 | 0 |
| T2 | 0 | ||
| T3 | 0.3 | ||
| T4 | 1.17 |
| Clone | Pathogenic Variants | Time Point | Proportion (%) |
|---|---|---|---|
| C1 | DNMT3AR882C TET2N1584Kfs*6 NPM1W288Cfs*12 FLT3-ITD3nt | T1 | 18.38 |
| T2 | 0 | ||
| T3 | 0 | ||
| C2 | DNMT3AR882C TET2N1584Kfs*6 NPM1W288Cfs*12 | T1 | 11.21 |
| T2 | 0 | ||
| T3 | 0 | ||
| C3 | DNMT3AR882C TET2N1584Kfs*6 NPM1W288Cfs*12 FLT3-ITD12nt | T1 | 4.58 |
| T2 | 0 | ||
| T3 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, S.; Borsi, E.; Patuelli, A.; Bandini, L.; Mancini, M.; Forte, D.; Nanni, J.; Barone, M.; Grassi, A.; Cristiano, G.; et al. Tracking Response and Resistance in Acute Myeloid Leukemia through Single-Cell DNA Sequencing Helps Uncover New Therapeutic Targets. Int. J. Mol. Sci. 2024, 25, 10002. https://doi.org/10.3390/ijms251810002
Bruno S, Borsi E, Patuelli A, Bandini L, Mancini M, Forte D, Nanni J, Barone M, Grassi A, Cristiano G, et al. Tracking Response and Resistance in Acute Myeloid Leukemia through Single-Cell DNA Sequencing Helps Uncover New Therapeutic Targets. International Journal of Molecular Sciences. 2024; 25(18):10002. https://doi.org/10.3390/ijms251810002
Chicago/Turabian StyleBruno, Samantha, Enrica Borsi, Agnese Patuelli, Lorenza Bandini, Manuela Mancini, Dorian Forte, Jacopo Nanni, Martina Barone, Alessandra Grassi, Gianluca Cristiano, and et al. 2024. "Tracking Response and Resistance in Acute Myeloid Leukemia through Single-Cell DNA Sequencing Helps Uncover New Therapeutic Targets" International Journal of Molecular Sciences 25, no. 18: 10002. https://doi.org/10.3390/ijms251810002