
Autophagy as a Guardian of Vascular Niche Homeostasis


{kind=link}
{kind=link}
Abstract
:1. Introduction
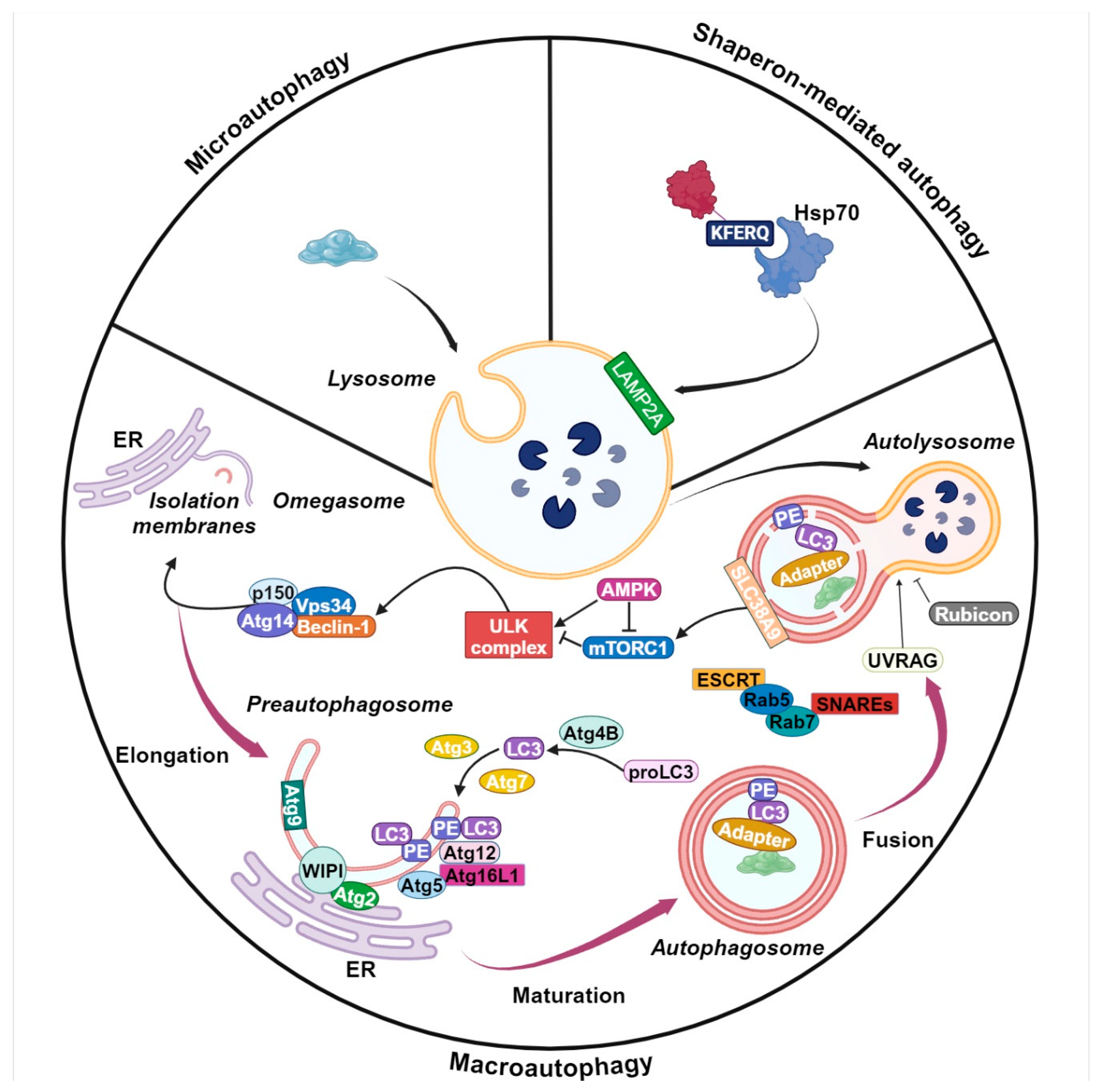
2. Autophagy Machinery
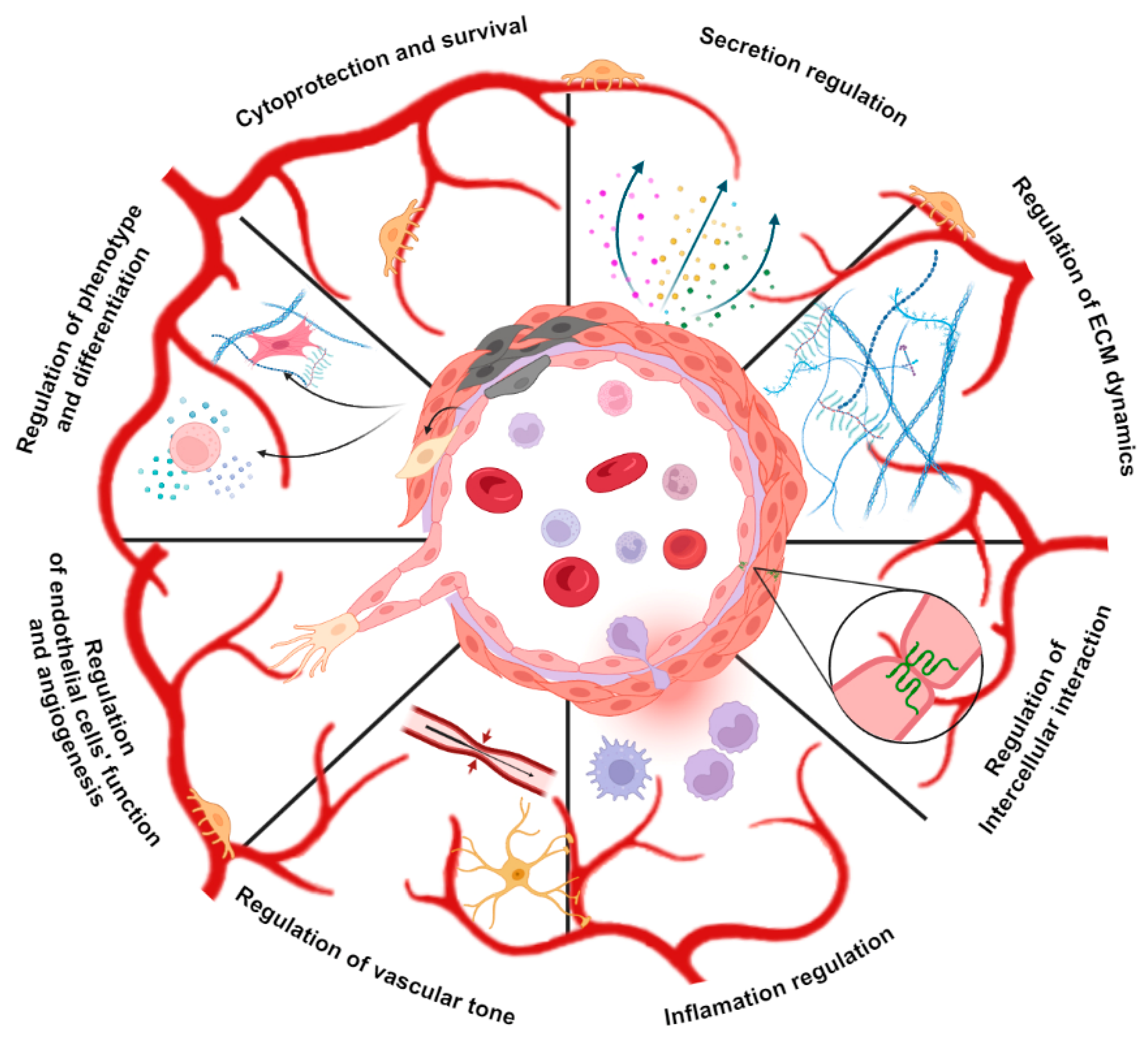
3. Autophagy Regulates Vascular Stem Cells
4. Autophagy Is a Switcher of Vascular Cells Phenotype
5. Autophagy Regulates Cell-Cell Interactions, Endothelial Barrier Stability and Inflammation State in Vascular Niches
6. Autophagy Regulates Cell-ECM Interactions in Perivascular Niche Cells
7. Autophagy Regulates Endothelial Cell Function and Angiogenesis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ribatti, D.; Tamma, R.; Annese, T. The Role of Vascular Niche and Endothelial Cells in Organogenesis and Regeneration. Exp. Cell Res. 2021, 398, 112398. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Hu, X.; Wu, Y.; Fu, L.; Lai, S.; Lin, J.; Li, X.; Lv, Y. The Role and Mechanism of the Vascular Endothelial Niche in Diseases: A Review. Front. Physiol. 2022, 13, 863265. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Xu, J.; Warren, C.M.; Duan, D.; Li, X.; Wu, L.; Iruela-Arispe, M.L. Endothelial Cells Provide an Instructive Niche for the Differentiation and Functional Polarization of M2-like Macrophages. Blood 2012, 120, 3152–3162. [Google Scholar] [CrossRef] [PubMed]
- Rouget, C. Memoire Sur Les Development, La Structure et La Proprietes Physiologiques Des Capillaires Sanguines et Lymphatiques. Arch. Physiol. 1873, 5, 603–663. [Google Scholar]
- Eberth, C.J. Handbuch Der Lehre von Der Gewegen Des Menschen und Der Tiereitle Bd.1; Verlag von Wilhelm Engelmann: Leipzig, Germany, 1871. [Google Scholar]
- Tilki, D.; Hohn, H.P.; Ergün, B.; Rafii, S.; Ergün, S. Emerging Biology of Vascular Wall Progenitor Cells in Health and Disease. Trends Mol. Med. 2009, 15, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gaviro, M.V.; Lovell-Badge, R.; Fernández-Avilés, F.; Lara-Pezzi, E. The Vascular Stem Cell Niche. J. Cardiovasc. Transl. Res. 2012, 5, 618–630. [Google Scholar] [CrossRef]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef]
- Corselli, M.; Chin, C.J.; Parekh, C.; Sahaghian, A.; Wang, W.; Ge, S.; Evseenko, D.; Wang, X.; Montelatici, E.; Lazzari, L.; et al. Perivascular Support of Human Hematopoietic Stem/Progenitor Cells. Blood 2013, 121, 2891–2901. [Google Scholar] [CrossRef]
- Chen, W.C.W.; Baily, J.E.; Corselli, M.; Díaz, M.E.; Sun, B.; Xiang, G.; Gray, G.A.; Huard, J.; Péault, B. Human Myocardial Pericytes: Multipotent Mesodermal Precursors Exhibiting Cardiac Specificity. Stem Cells 2015, 33, 557–573. [Google Scholar] [CrossRef]
- Peisker, F.; Halder, M.; Nagai, J.; Ziegler, S.; Kaesler, N.; Hoeft, K.; Li, R.; Bindels, E.M.J.; Kuppe, C.; Moellmann, J.; et al. Mapping the Cardiac Vascular Niche in Heart Failure. Nat. Commun. 2022, 13, 3027. [Google Scholar] [CrossRef]
- Oh, M.; Nör, J.E. The Perivascular Niche and Self-Renewal of Stem Cells. Front. Physiol. 2015, 6, 367. [Google Scholar] [CrossRef] [PubMed]
- Cardier, J.E.; Barberá-Guillem, E. Extramedullary Hematopoiesis in the Adult Mouse Liver Is Associated with Specific Hepatic Sinusoidal Endothelial Cells. Hepatology 1997, 26, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Johnson, S.A.; Shelley, W.C.; Yoder, M.C. Hematopoietic Stem Cell Repopulating Ability Can Be Maintained in Vitro by Some Primary Endothelial Cells. Exp. Hematol. 2004, 32, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Yilmaz, Ö.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and Perivascular Cells Maintain Haematopoietic Stem Cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef]
- Bourke, V.A.; Watchman, C.J.; Reith, J.D.; Jorgensen, M.L.; Dieudonnè, A.; Bolch, W.E. Spatial Gradients of Blood Vessels and Hematopoietic Stem and Progenitor Cells within the Marrow Cavities of the Human Skeleton. Blood 2009, 114, 4077–4080. [Google Scholar] [CrossRef]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar Niches Maintain Haematopoietic Stem Cell Quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef]
- Kiel, M.J.; Morrison, S.J. Uncertainty in the Niches That Maintain Haematopoietic Stem Cells. Nat. Rev. Immunol. 2008, 8, 290–301. [Google Scholar] [CrossRef]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult Haematopoietic Stem Cell Niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef]
- Nombela-Arrieta, C.; Pivarnik, G.; Winkel, B.; Canty, K.J.; Harley, B.; Mahoney, J.E.; Park, S.Y.; Lu, J.; Protopopov, A.; Silberstein, L.E. Quantitative Imaging of Haematopoietic Stem and Progenitor Cell Localization and Hypoxic Status in the Bone Marrow Microenvironment. Nat. Cell Biol. 2013, 15, 533–543. [Google Scholar] [CrossRef]
- Tavazoie, M.; Van der Veken, L.; Silva-Vargas, V.; Louissaint, M.; Colonna, L.; Zaidi, B.; Garcia-Verdugo, J.M.; Doetsch, F. A Specialized Vascular Niche for Adult Neural Stem Cells. Cell Stem Cell 2008, 3, 279–288. [Google Scholar] [CrossRef]
- Mammoto, A.; Mammoto, T. Vascular Niche in Lung Alveolar Development, Homeostasis, and Regeneration. Front. Bioeng. Biotechnol. 2019, 7, 475993. [Google Scholar] [CrossRef] [PubMed]
- Peña-Jimenez, D.; Fontenete, S.; Megias, D.; Fustero-Torre, C.; Graña-Castro, O.; Castellana, D.; Loewe, R.; Perez-Moreno, M. Lymphatic Vessels Interact Dynamically with the Hair Follicle Stem Cell Niche during Skin Regeneration In Vivo. EMBO J. 2019, 38, 2. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, F.; Longo, G.; Vancheri, S.; Henein, M. Coronary Microvascular Dysfunction. J. Clin. Med. 2020, 9, 2880. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, H.; Ranek, M.J. Protein Quality Control and Degradation in Cardiomyocytes. J. Mol. Cell. Cardiol. 2008, 45, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Beau, I.; Esclatine, A.; Codogno, P. Lost to Translation: When Autophagy Targets Mature Ribosomes. Trends Cell Biol. 2008, 18, 311–314. [Google Scholar] [CrossRef]
- Majeski, A.E.; Fred Dice, J. Mechanisms of Chaperone-Mediated Autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444. [Google Scholar] [CrossRef]
- Ravikumar, B.; Futter, M.; Jahreiss, L.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; Massey, D.C.O.; Menzies, F.M.; Narayanan, U.; Renna, M.; et al. Mammalian Macroautophagy at a Glance. J. Cell Sci. 2009, 122, 1707–1711. [Google Scholar] [CrossRef]
- Suzuki, K.; Kubota, Y.; Sekito, T.; Ohsumi, Y. Hierarchy of Atg Proteins in Pre-Autophagosomal Structure Organization. Genes Cells 2007, 12, 209–218. [Google Scholar] [CrossRef]
- Pattingre, S.; Espert, L.; Biard-Piechaczyk, M.; Codogno, P. Regulation of Macroautophagy by MTOR and Beclin 1 Complexes. Biochimie 2008, 90, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Mechanisms of Selective Autophagy. Annu. Rev. Cell Dev. Biol. 2021, 37, 143–169. [Google Scholar] [CrossRef] [PubMed]
- Faruk, M.O.; Ichimura, Y.; Komatsu, M. Selective Autophagy. Cancer Sci. 2021, 112, 3972–3978. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Yoshimori, T. A Current Perspective of Autophagosome Biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Turco, E.; Witt, M.; Abert, C.; Bock-Bierbaum, T.; Su, M.Y.; Trapannone, R.; Sztacho, M.; Danieli, A.; Shi, X.; Zaffagnini, G.; et al. FIP200 Claw Domain Binding to P62 Promotes Autophagosome Formation at Ubiquitin Condensates. Mol. Cell 2019, 74, 330–346.e11. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 347–362.e6. [Google Scholar] [CrossRef]
- Ravenhill, B.J.; Boyle, K.B.; von Muhlinen, N.; Ellison, C.J.; Masson, G.R.; Otten, E.G.; Foeglein, A.; Williams, R.; Randow, F. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 2019, 74, 320–329.e6. [Google Scholar] [CrossRef]
- Li, J.; McCullough, L.D. Effects of AMP-Activated Protein Kinase in Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 480–492. [Google Scholar] [CrossRef]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network Organization of the Human Autophagy System. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef]
- Simonsen, A.; Tooze, S.A. Coordination of Membrane Events during Autophagy by Multiple Class III PI3-Kinase Complexes. J. Cell Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and Diversity in Autophagy Mechanisms: Lessons from Yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Matsushita, M.; Suzuki, N.N.; Obara, K.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structure of Atg5·Atg16, a Complex Essential for Autophagy. J. Biol. Chem. 2007, 282, 6763–6772. [Google Scholar] [CrossRef] [PubMed]
- Lőrincz, P.; Juhász, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.S.; Liton, P.B. The Physiological and Pathophysiological Roles of the Autophagy Lysosomal System in the Conventional Aqueous Humor Outflow Pathway: More than Cellular Clean Up. Prog. Retin. Eye Res. 2022, 90, 101064. [Google Scholar] [CrossRef] [PubMed]
- Ewald, M.L.; Chen, Y.H.; Lee, A.P.; Hughes, C.C.W. The Vascular Niche in next Generation Microphysiological Systems. Lab Chip 2021, 21, 3244–3262. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef]
- Bearzi, C.; Rota, M.; Hosoda, T.; Tillmanns, J.; Nascimbene, A.; De Angelis, A.; Yasuzawa-Amano, S.; Trofimova, I.; Siggins, R.W.; LeCapitaine, N.; et al. Human Cardiac Stem Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 14068–14073. [Google Scholar] [CrossRef]
- Bautch, V.L. Stem Cells and the Vasculature. Nat. Med. 2011, 17, 1437–1443. [Google Scholar] [CrossRef]
- Beltrami, A.P.; Madeddu, P. Pericytes and Cardiac Stem Cells: Common Features and Peculiarities. Pharmacol. Res. 2018, 127, 101–109. [Google Scholar] [CrossRef]
- Herrero, D.; Albericio, G.; Higuera, M.; Herranz-López, M.; García-Brenes, M.A.; Cordero, A.; Roche, E.; Sepúlveda, P.; Mora, C.; Bernad, A. The Vascular Niche for Adult Cardiac Progenitor Cells. Antioxidants 2022, 11, 882. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dasari, C.; Li, D.; Arshia, A.; Umer, A.M.; Abouzid, M.R.A.; Guo, Y.; Bolli, R. Effects of Heme Oxygenase-1 on c-Kit-Positive Cardiac Cells. Int. J. Mol. Sci. 2021, 22, 13448. [Google Scholar] [CrossRef] [PubMed]
- Korski, K.I.; Kubli, D.A.; Wang, B.J.; Khalafalla, F.G.; Monsanto, M.M.; Firouzi, F.; Echeagaray, O.H.; Kim, T.; Adamson, R.M.; Dembitsky, W.P.; et al. Hypoxia Prevents Mitochondrial Dysfunction and Senescence in Human C-Kit+ Cardiac Progenitor Cells. Stem Cells 2019, 37, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Bergeron, D.L.; Celeste Simon, M.; Ramírez-bergeron, D.L. Hypoxia-Inducible Factor and the Development of Stem Cells of the Cardiovascular System. Stem Cells 2001, 19, 279–286. [Google Scholar] [CrossRef]
- Kimura, W.; Sadek, H.A. The Cardiac Hypoxic Niche: Emerging Role of Hypoxic Microenvironment in Cardiac Progenitors. Cardiovasc. Diagn. Ther. 2012, 2, 278. [Google Scholar] [CrossRef] [PubMed]
- Moscoso, I.; Tejados, N.; Barreiro, O.; Sepúlveda, P.; Izarra, A.; Calvo, E.; Dorronsoro, A.; Salcedo, J.M.; Sádaba, R.; Díez-Juan, A.; et al. Podocalyxin-like Protein 1 Is a Relevant Marker for Human c-kitpos Cardiac Stem Cells. J. Tissue Eng. Regen. Med. 2016, 10, 580–590. [Google Scholar] [CrossRef]
- van Oorschot, A.A.M.; Smits, A.M.; Pardali, E.; Doevendans, P.A.; Goumans, M.J. Low Oxygen Tension Positively Influences Cardiomyocyte Progenitor Cell Function. J. Cell. Mol. Med. 2011, 15, 2723–2734. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, X.X.; Li, J.Z.; Qiang, H.F.; Wang, Y.; Li, G. Pretreatment of Cardiac Progenitor Cells with Bradykinin Attenuates H2O2-Induced Cell Apoptosis and Improves Cardiac Function in Rats by Regulating Autophagy. Stem Cell Res. Ther. 2021, 12, 437. [Google Scholar] [CrossRef]
- Ma, W.; Ding, F.; Wang, X.; Huang, Q.; Zhang, L.; Bi, C.; Hua, B.; Yuan, Y.; Han, Z.; Jin, M.; et al. By Targeting Atg7 MicroRNA-143 Mediates Oxidative Stress-Induced Autophagy of c-Kit+ Mouse Cardiac Progenitor Cells. EBioMedicine 2018, 32, 182–191. [Google Scholar] [CrossRef]
- Shi, X.; Li, W.; Liu, H.; Yin, D.; Zhao, J. The ROS/NF-ΚB/NR4A2 Pathway Is Involved in H2O2 Induced Apoptosis of Resident Cardiac Stem Cells via Autophagy. Oncotarget 2017, 8, 77634–77648. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, A.V.; Eapen, M.S.; McAlinden, K.D.; Chia, C.; Larby, J.; Myers, S.; Dey, S.; Haug, G.; Markos, J.; Glanville, A.R.; et al. Endothelial to Mesenchymal Transition (EndMT) and Vascular Remodeling in Pulmonary Hypertension and Idiopathic Pulmonary Fibrosis. Expert Rev. Respir. Med. 2020, 14, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, Y.; Lee, S.M.; Dongqing, Z.; Kitada, M.; Kanasaki, K.; Koya, D. Endothelial Autophagy Deficiency Induces IL6—Dependent Endothelial Mesenchymal Transition and Organ Fibrosis. Autophagy 2020, 16, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Tian, K.J.; Yan, B.J.; Gui, D.D.; Luo, W.; Ren, Z.; Wei, D.H.; Liu, L.S.; Jiang, Z.S. A Promising Field: Regulating Imbalance of EndMT in Cardiovascular Diseases. Cell Cycle 2021, 20, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Kong, S.; Dong, T.; Lin, Z.; Zhang, Q.; Chen, X.; Gong, Y.; Fan, X.; He, M.; Zhou, H. Autophagy Modulates Mesenchymal-to-Endothelial Transition via P53. Aging 2020, 12, 22112–22121. [Google Scholar] [CrossRef]
- Zhang, D.; Cao, Y.; Liu, D.; Zhang, J.; Guo, Y. The Etiology and Molecular Mechanism Underlying Smooth Muscle Phenotype Switching in Intimal Hyperplasia of Vein Graft and the Regulatory Role of MicroRNAs. Front. Cardiovasc. Med. 2022, 9, 935054. [Google Scholar] [CrossRef]
- MacK, C.P. Signaling Mechanisms That Regulate Smooth Muscle Cell Differentiation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef]
- Salabei, J.K.; Cummins, T.D.; Singh, M.; Jones, S.P.; Bhatnagar, A.; Hill, B.G. PDGF-Mediated Autophagy Regulates Vascular Smooth Muscle Cell Phenotype and Resistance to Oxidative Stress. Biochem. J. 2013, 451, 375–388. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, J.; Ye, L.C.; Li, J.; Wu, Z.; Li, Y.; Li, C. Autophagy Maintains the Integrity of Endothelial Barrier in LPS-Induced Lung Injury. J. Cell. Physiol. 2018, 233, 688–698. [Google Scholar] [CrossRef]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 Controls Endothelial Adherens Junctions, Cell-Cell Tension, Angiogenesis, and Barrier Formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef]
- Li, H.; Gao, A.; Feng, D.; Wang, Y.; Zhang, L.; Cui, Y.; Li, B.; Wang, Z.; Chen, G. Evaluation of the Protective Potential of Brain Microvascular Endothelial Cell Autophagy on Blood-Brain Barrier Integrity During Experimental Cerebral Ischemia-Reperfusion Injury. Transl. Stroke Res. 2014, 5, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Ding, J.; Zhou, X.; Chen, G.; Shu, F.L. Divergent Roles of Endothelial NF-KappaB in Multiple Organ Injury and Bacterial Clearance in Mouse Models of Sepsis. J. Exp. Med. 2008, 205, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Fazal, F. Hug Tightly and Say Goodbye: Role of Endothelial ICAM-1 in Leukocyte Transmigration. Antioxid. Redox Signal. 2009, 11, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Fazal, F.; Bijli, K.M.; Minhajuddin, M.; Rein, T.; Finkelstein, J.N.; Rahman, A. Essential Role of Cofilin-1 in Regulating Thrombin-Induced RelA/P65 Nuclear Translocation and Intercellular Adhesion Molecule 1 (ICAM-1) Expression in Endothelial Cells. J. Biol. Chem. 2009, 284, 21047–21056. [Google Scholar] [CrossRef] [PubMed]
- Minhajuddin, M.; Bijli, K.M.; Fazal, F.; Sassano, A.; Nakayama, K.I.; Hay, N.; Platanias, L.C.; Rahman, A. Protein Kinase C-Delta and Phosphatidylinositol 3-Kinase/Akt Activate Mammalian Target of Rapamycin to Modulate NF-KappaB Activation and Intercellular Adhesion Molecule-1 (ICAM-1) Expression in Endothelial Cells. J. Biol. Chem. 2009, 284, 4052–4061. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Peng, W.; Lin, X.; Huang, Q.L.; Lin, J.Y. PLC/CAMK IV-NF-KappaB Involved in the Receptor for Advanced Glycation End Products Mediated Signaling Pathway in Human Endothelial Cells. Mol. Cell. Endocrinol. 2010, 320, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Paria, B.C.; Malik, A.B.; Kwiatek, A.M.; Rahman, A.; May, M.J.; Ghosh, S.; Tiruppathi, C. Tumor Necrosis Factor-Alpha Induces Nuclear Factor-KappaB-Dependent TRPC1 Expression in Endothelial Cells. J. Biol. Chem. 2003, 278, 37195–37203. [Google Scholar] [CrossRef]
- Leonard, A.; Millar, M.W.; Slavin, S.A.; Bijli, K.M.; Dionisio Santos, D.A.; Dean, D.A.; Fazal, F.; Rahman, A. Critical Role of Autophagy Regulator Beclin1 in Endothelial Cell Inflammation and Barrier Disruption. Cell. Signal. 2019, 61, 120–129. [Google Scholar] [CrossRef]
- Yoshimura, A.; Perretti, M.; Aksoy, E.; Correspondence, S.N.; Reglero-Real, N.; Pé Rez-Gutié Rrez, L.; Rolas, L.L.; Garrido-Mesa, J.; Barkaway, A.; Pickworth, C.; et al. Autophagy Modulates Endothelial Junctions to Restrain Neutrophil Diapedesis during Inflammation. Immunity 2021, 54, 1989–2004.e9. [Google Scholar] [CrossRef]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- Reglero-Real, N.; Colom, B.; Bodkin, J.V.; Nourshargh, S. Endothelial Cell Junctional Adhesion Molecules. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. How Leukocytes Cross the Vascular Endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Crişan, T.O.; Plantinga, T.S.; van de Veerdonk, F.L.; Farcaş, M.F.; Stoffels, M.; Kullberg, B.J.; van der Meer, J.W.M.; Joosten, L.A.B.; Netea, M.G. Inflammasome-Independent Modulation of Cytokine Response by Autophagy in Human Cells. PLoS ONE 2011, 6, e18666. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy Controls IL-1beta Secretion by Targeting pro-IL-1beta for Degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–226. [Google Scholar] [CrossRef]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-Based Unconventional Secretory Pathway for Extracellular Delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef]
- Zhuang, S.F.; Liu, D.X.; Wang, H.J.; Zhang, S.H.; Wei, J.Y.; Fang, W.G.; Zhang, K.; Cao, L.; Zhao, W.D.; Chen, Y.H. Atg7 Regulates Brain Angiogenesis via NF-ΚB-Dependent IL-6 Production. Int. J. Mol. Sci. 2017, 18, 968. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Shtengel, G.; Pasapera, A.M.; Ramko, E.B.; Davidson, M.W.; Hess, H.F.; Waterman, C.M. Nanoscale Architecture of Integrin-Based Cell Adhesions. Nat. 2010, 468, 580–584. [Google Scholar] [CrossRef]
- Swiatlowska, P.; Iskratsch, T. Tools for Studying and Modulating (Cardiac Muscle) Cell Mechanics and Mechanosensing across the Scales. Biophys. Rev. 2021, 13, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Yim, E.K.F.; Kanchanawong, P. Molecular Organization of Integrin-Based Adhesion Complexes in Mouse Embryonic Stem Cells. ACS Biomater. Sci. Eng. 2019, 5, 3828–3842. [Google Scholar] [CrossRef] [PubMed]
- Vlahakis, A.; Debnath, J. The Interconnections between Autophagy and Integrin-Mediated Cell Adhesion. J. Mol. Biol. 2017, 429, 515–530. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016, 15, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Yamada, E.; Okada, S.; Bastie, C.C.; Vatish, M.; Nakajima, Y.; Shibusawa, R.; Ozawa, A.; Pessin, J.E.; Yamada, M. Fyn Phosphorylates AMPK to Inhibit AMPK Activity and AMP-Dependent Activation of Autophagy. Oncotarget 2016, 7, 74612–74629. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.C.; Lee, J.Y.; Tang, H.W.; Debnath, J.; Thomas, S.M.; Settleman, J. Genetic Interactions between Drosophila Melanogaster Atg1 and Paxillin Reveal a Role for Paxillin in Autophagosome Formation. Autophagy 2008, 4, 37–45. [Google Scholar] [CrossRef]
- Zhao, M.; Vuori, K. The Docking Protein P130Cas Regulates Cell Sensitivity to Proteasome Inhibition. BMC Biol. 2011, 9, 73. [Google Scholar] [CrossRef]
- Buraschi, S.; Neill, T.; Goyal, A.; Poluzzi, C.; Smythies, J.; Owens, R.T.; Schaefer, L.; Torres, A.; Iozzo, R.V. Decorin Causes Autophagy in Endothelial Cells via Peg3. Proc. Natl. Acad. Sci. USA 2013, 110, E2582–E2591. [Google Scholar] [CrossRef]
- Neill, T.; Torres, A.; Buraschi, S.; Iozzo, R.V. Decorin Has an Appetite for Endothelial Cell Autophagy. Autophagy 2013, 9, 1626–1628. [Google Scholar] [CrossRef]
- Goyal, A.; Poluzzi, C.; Willis, C.D.; Smythies, J.; Shellard, A.; Neill, T.; Iozzo, R.V. Endorepellin Affects Angiogenesis by Antagonizing Diverse Vascular Endothelial Growth Factor Receptor 2 (VEGFR2)-Evoked Signaling Pathways: Transcriptional Repression of Hypoxia-Inducible Factor 1α and VEGFA and Concurrent Inhibition of Nuclear Factor of Activated T Cell 1 (NFAT1) Activation. J. Biol. Chem. 2012, 287, 43543–43556. [Google Scholar] [CrossRef]
- Nguyen, T.M.B.; Subramanian, I.V.; Kelekar, A.; Ramakrishnan, S. Kringle 5 of Human Plasminogen, an Angiogenesis Inhibitor, Induces Both Autophagy and Apoptotic Death in Endothelial Cells. Blood 2007, 109, 4793–4802. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Ji, Y.; Chen, Z.; Kitazato, K.; Xiang, Y.; Zhong, M.; Wang, Q.; Pei, Y.; Ju, H.; Wang, Y. Proteomics Analysis of Autophagy-Deficient Atg7−/− MEFs Reveals a Close Relationship between F-Actin and Autophagy. Biochem. Biophys. Res. Commun. 2013, 437, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Cui, L.H.; Zhuo, Y.Z.; Hu, J.G.; Cui, N.; Zhang, S.K. Inhibiting Autophagy Promotes Collagen Degradation by Regulating Matrix Metalloproteinases in Pancreatic Stellate Cells. Life Sci. 2018, 208, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Keulers, T.G.; Schaaf, M.B.E.; Rouschop, K.M.A. Autophagy-Dependent Secretion: Contribution to Tumor Progression. Front. Oncol. 2016, 6, 233818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; An, Q.; Wang, T.; Gao, S.; Zhou, G. Autophagy- and MMP-2/9-Mediated Reduction and Redistribution of ZO-1 Contribute to Hyperglycemia-Increased Blood–Brain Barrier Permeability During Early Reperfusion in Stroke. Neuroscience 2018, 377, 126–137. [Google Scholar] [CrossRef]
- Oh, S.; Hwang, J.R.; Choi, M.; Kim, Y.M.; Kim, J.S.; Suh, Y.L.; Choi, S.J.; Roh, C.R. Autophagy Regulates Trophoblast Invasion by Targeting NF-ΚB Activity. Sci. Rep. 2020, 10, 14033. [Google Scholar] [CrossRef]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-Dependent Production of Secreted Factors Facilitates Oncogenic RAS-Driven Invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef]
- Öhman, T.; Teirilä, L.; Lahesmaa-Korpinen, A.-M.; Cypryk, W.; Veckman, V.; Saijo, S.; Wolff, H.; Hautaniemi, S.; Nyman, T.A.; Matikainen, S. Dectin-1 Pathway Activates Robust Autophagy-Dependent Unconventional Protein Secretion in Human Macrophages. J. Immunol. 2014, 192, 5952–5962. [Google Scholar] [CrossRef]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated SNAREs and Specialized TRIM Cargo Receptors Mediate Secretory Autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef]
- Lin, L.; Gao, W.; Feng, L.; Wang, C.; Yang, R.; Wang, W.; Wu, Q. Autophagy Induced by Low Shear Stress Leads to Endothelial Glycocalyx Disruption. J. Vasc. Res. 2024, 61, 77–88. [Google Scholar] [CrossRef]
- Spengler, K.; Kryeziu, N.; Große, S.; Mosig, A.S.; Heller, R. VEGF Triggers Transient Induction of Autophagy in Endothelial Cells via AMPKα1. Cells 2020, 9, 687. [Google Scholar] [CrossRef] [PubMed]
- Stahmann, N.; Woods, A.; Spengler, K.; Heslegrave, A.; Bauer, R.; Krause, S.; Viollet, B.; Carling, D.; Heller, R. Activation of AMP-Activated Protein Kinase by Vascular Endothelial Growth Factor Mediates Endothelial Angiogenesis Independently of Nitric-Oxide Synthase. J. Biol. Chem. 2010, 285, 10638–10652. [Google Scholar] [CrossRef] [PubMed]
- Nagata, D.; Mogi, M.; Walsh, K. AMP-Activated Protein Kinase (AMPK) Signaling in Endothelial Cells Is Essential for Angiogenesis in Response to Hypoxic Stress. J. Biol. Chem. 2003, 278, 31000–31006. [Google Scholar] [CrossRef] [PubMed]
- Reihill, J.A.; Ewart, M.A.; Salt, I.P. The Role of AMP-Activated Protein Kinase in the Functional Effects of Vascular Endothelial Growth Factor-A and -B in Human Aortic Endothelial Cells. Vasc. Cell 2011, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.P.; Mitchelhill, K.I.; Michell, B.J.; Stapleton, D.; Rodriguez-Crespo, I.; Witters, L.A.; Power, D.A.; Ortiz De Montellano, P.R.; Kemp, B.E. AMP-Activated Protein Kinase Phosphorylation of Endothelial NO Synthase. FEBS Lett. 1999, 443, 285–289. [Google Scholar] [CrossRef]
- Mount, P.F.; Hill, R.E.; Fraser, S.A.; Levidiotis, V.; Katsis, F.; Kemp, B.E.; Power, D.A. Acute Renal Ischemia Rapidly Activates the Energy Sensor AMPK but Does Not Increase Phosphorylation of ENOS-Ser1177. Am. J. Physiol. Renal Physiol. 2005, 289, F1103–F1115. [Google Scholar] [CrossRef]
- Morrow, V.A.; Foufelle, F.; Connell, J.M.C.; Petrie, J.R.; Gould, G.W.; Salt, I.P. Direct Activation of AMP-Activated Protein Kinase Stimulates Nitric-Oxide Synthesis in Human Aortic Endothelial Cells. J. Biol. Chem. 2003, 278, 31629–31639. [Google Scholar] [CrossRef]
- Stahmann, N.; Woods, A.; Carling, D.; Heller, R. Thrombin Activates AMP-Activated Protein Kinase in Endothelial Cells via a Pathway Involving Ca2+/Calmodulin-Dependent Protein Kinase Kinase Beta. Mol. Cell. Biol. 2006, 26, 5933–5945. [Google Scholar] [CrossRef]
- Chen, Z.; Peng, I.C.; Sun, W.; Su, M.I.; Hsu, P.H.; Fu, Y.; Zhu, Y.; Defea, K.; Pan, S.; Tsai, M.D.; et al. AMP-Activated Protein Kinase Functionally Phosphorylates Endothelial Nitric Oxide Synthase Ser633. Circ. Res. 2009, 104, 496–505. [Google Scholar] [CrossRef]
- Bharath, L.P.; Mueller, R.; Li, Y.; Ruan, T.; Kunz, D.; Goodrich, R.; Mills, T.; Deeter, L.; Sargsyan, A.; Babu, P.V.A.; et al. Impairment of Autophagy in Endothelial Cells Prevents Shear-Stress-Induced Increases in Nitric Oxide Bioavailability. Can. J. Physiol. Pharmacol. 2014, 92, 605–612. [Google Scholar] [CrossRef]
- McCarthy, C.G.; Wenceslau, C.F.; Calmasini, F.B.; Klee, N.S.; Brands, M.W.; Joe, B.; Webb, R.C. Reconstitution of Autophagy Ameliorates Vascular Function and Arterial Stiffening in Spontaneously Hypertensive Rats. Am. J. Physiol.-Heart Circ. Physiol. 2019, 317, H1013–H1027. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Kwon, Y.; Byeon, S.; Lee, Y.H. Stimulation of Autophagy Improves Vascular Function in the Mesenteric Arteries of Type 2 Diabetic Mice. Exp. Physiol. 2020, 105, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Hughes, W.E.; Beyer, A.M.; Gutterman, D.D. Vascular Autophagy in Health and Disease. Basic Res. Cardiol. 2020, 115, 41. [Google Scholar] [CrossRef] [PubMed]
- Fetterman, J.L.; Holbrook, M.; Flint, N.; Feng, B.; Bret’n-Romero, R.; Linder, E.A.; Berk, B.D.; Duess, M.A.; Farb, M.G.; Gokce, N.; et al. Restoration of Autophagy in Endothelial Cells from Patients with Diabetes Mellitus Improves Nitric Oxide Signaling. Atherosclerosis 2016, 247, 207–217. [Google Scholar] [CrossRef]
- Nivoit, P.; Mathivet, T.; Wu, J.; Salemkour, Y.; Sankar, D.S.; Baudrie, V.; Bourreau, J.; Guihot, A.L.; Vessieres, E.; Lemitre, M.; et al. Autophagy Protein 5 Controls Flow-Dependent Endothelial Functions. Cell. Mol. Life Sci. 2023, 80, 210. [Google Scholar] [CrossRef]
- Domigan, C.K.; Warren, C.M.; Antanesian, V.; Happel, K.; Ziyad, S.; Lee, S.; Krall, A.; Duan, L.; Torres-Collado, A.X.; Castellani, L.W.; et al. Autocrine VEGF Maintains Endothelial Survival through Regulation of Metabolism and Autophagy. J. Cell Sci. 2015, 128, 2236–2248. [Google Scholar] [CrossRef]
- Zeng, M.; Wei, X.; Wu, Z.; Li, W.; Zheng, Y.; Li, B.; Meng, X.; Fu, X.; Fei, Y. Simulated Ischemia/Reperfusion-Induced P65-Beclin 1-Dependent Autophagic Cell Death in Human Umbilical Vein Endothelial Cells. Sci. Rep. 2016, 6, 37448. [Google Scholar] [CrossRef]
- Lin, H.H. In Vitro and in Vivo Atheroprotective Effects of Gossypetin against Endothelial Cell Injury by Induction of Autophagy. Chem. Res. Toxicol. 2016, 28, 202–215. [Google Scholar] [CrossRef]
- Luo, Z.; Yao, J.; Wang, Z.; Xu, J. Mitochondria in Endothelial Cells Angiogenesis and Function: Current Understanding and Future Perspectives. J. Transl. Med. 2023, 21, 441. [Google Scholar] [CrossRef]
- Wu, W.; Xu, H.; Wang, Z.; Mao, Y.; Yuan, Y.; Luo, W.; Cui, Z.; Cui, T.; Wang, X.L.; Shen, Y.H. PINK1-Parkin-Mediated Mitophagy Protects Mitochondrial Integrity and Prevents Metabolic Stress-Induced Endothelial Injury. PLoS ONE 2015, 10, e0132499. [Google Scholar] [CrossRef]
- Mameli, E.; Martello, A.; Caporali, A. Autophagy at the Interface of Endothelial Cell Homeostasis and Vascular Disease. FEBS J. 2022, 289, 2976–2991. [Google Scholar] [CrossRef] [PubMed]
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy Is Required for Endothelial Cell Alignment and Atheroprotection under Physiological Blood Flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Kim, H.H.; Hiroi, Y.; Sawada, N.; Salomone, S.; Benjamin, L.E.; Walsh, K.; Moskowitz, M.A.; Liao, J.K. Obesity Increases Vascular Senescence and Susceptibility to Ischemic Injury through Chronic Activation of Akt and MTOR. Sci. Signal. 2009, 2, ra11. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dergilev, K.; Gureenkov, A.; Parfyonova, Y. Autophagy as a Guardian of Vascular Niche Homeostasis. Int. J. Mol. Sci. 2024, 25, 10097. https://doi.org/10.3390/ijms251810097
Dergilev K, Gureenkov A, Parfyonova Y. Autophagy as a Guardian of Vascular Niche Homeostasis. International Journal of Molecular Sciences. 2024; 25(18):10097. https://doi.org/10.3390/ijms251810097
Chicago/Turabian StyleDergilev, Konstantin, Alexandre Gureenkov, and Yelena Parfyonova. 2024. "Autophagy as a Guardian of Vascular Niche Homeostasis" International Journal of Molecular Sciences 25, no. 18: 10097. https://doi.org/10.3390/ijms251810097