
RAMP1 Signaling Mitigates Acute Lung Injury by Distinctively Regulating Alveolar and Monocyte-Derived Macrophages
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
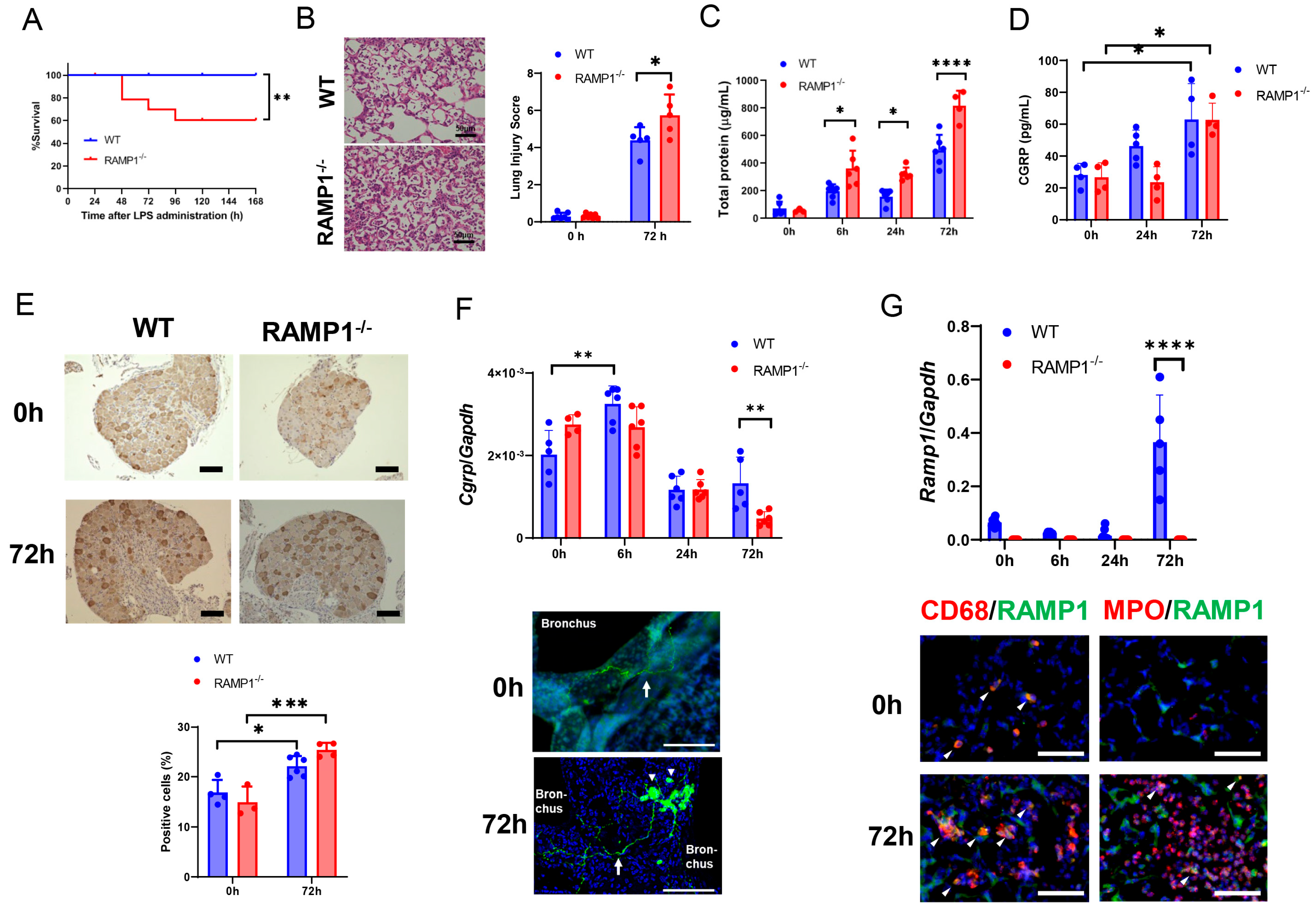
2.1. RAMP1 Signaling Deficiency Increases Mortality, Lung Injury, and Pulmonary Edema in LPS-Induced Acute Lung Injury
2.2. Expression of CGRP/RAMP1 during Acute Lung Injury
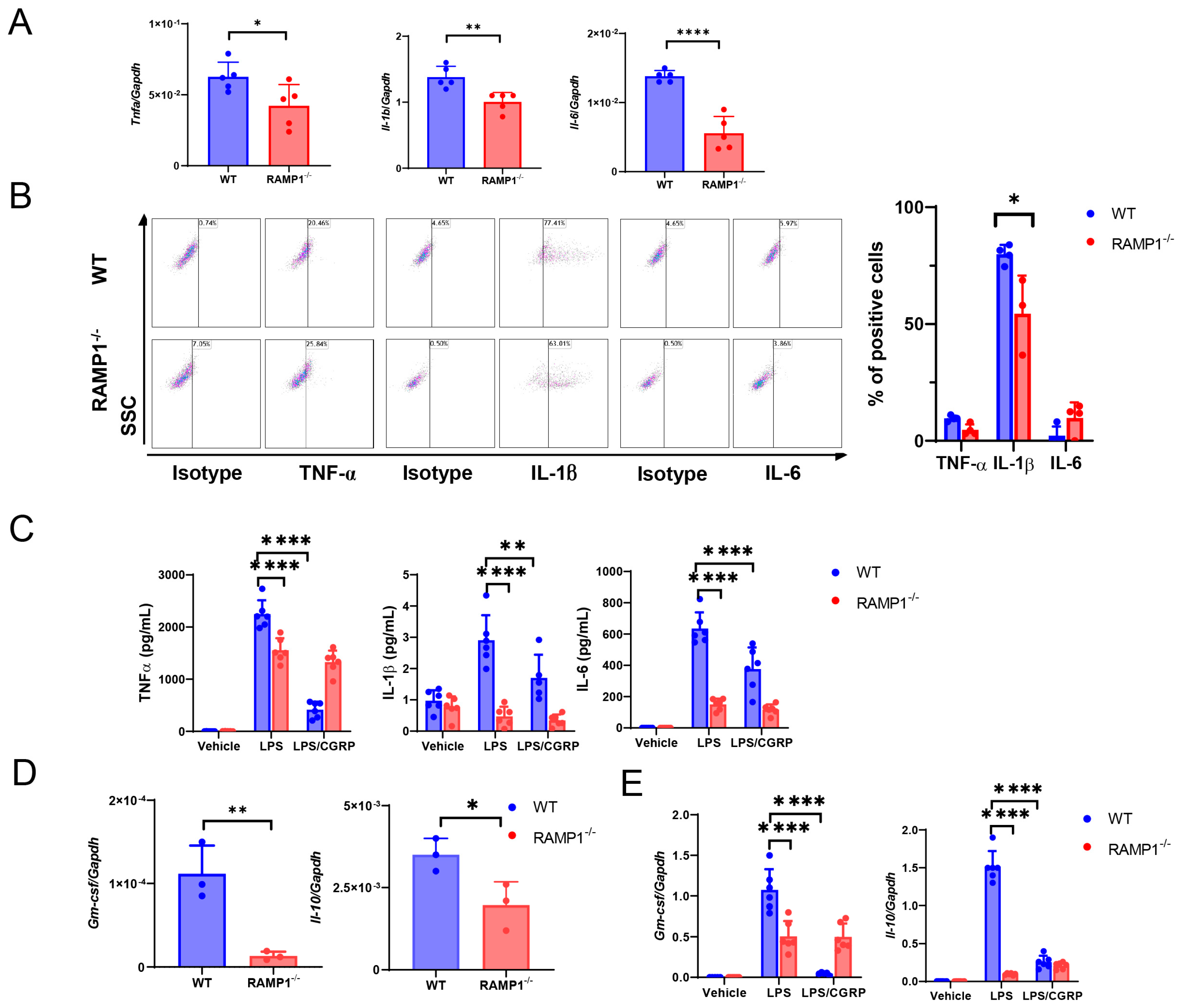
2.3. RAMP1 Signaling Deficiency Increases Cytokine Production
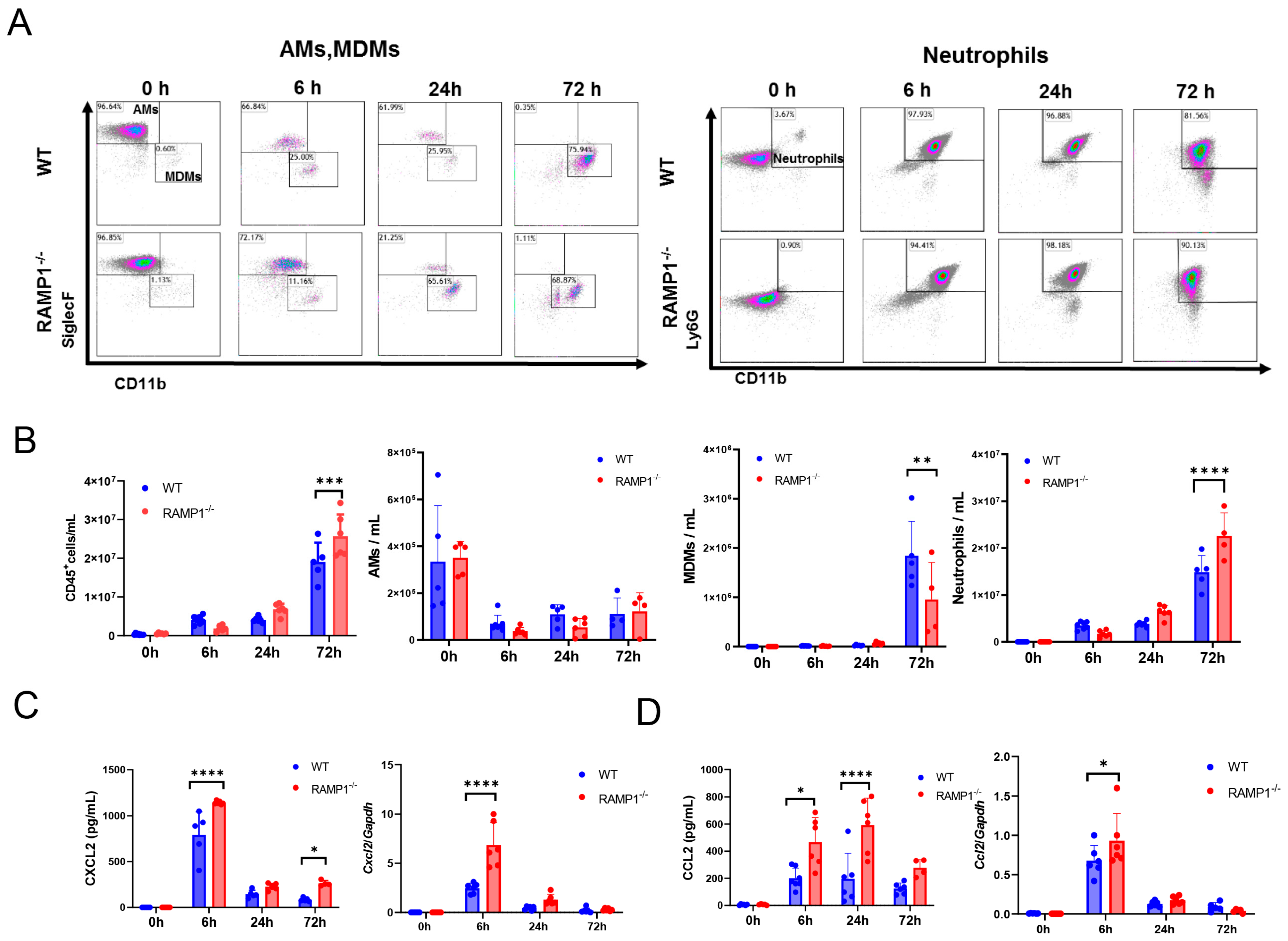
2.4. RAMP1 Signaling Deficiency Enhances Neutrophil Accumulation and Attenuates MDM Accumulation
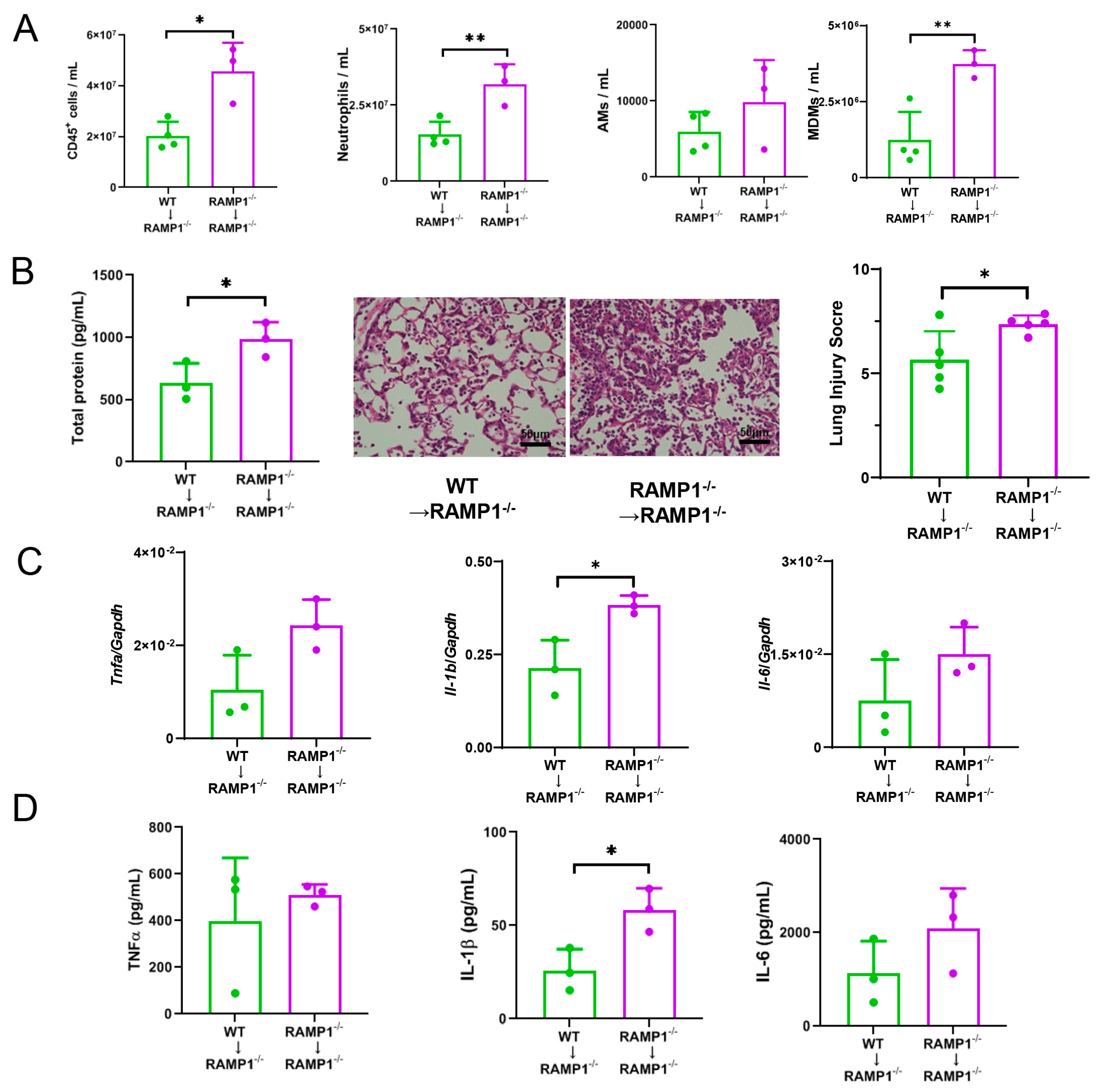
2.5. Roles of AMs in RAMP1 Signaling during ALI
2.6. MDMs in RAMP1 Signaling Contribute to LPS-Induced ALI
2.7. RAMP1 Signaling Suppresses Accumulation of Neutrophils
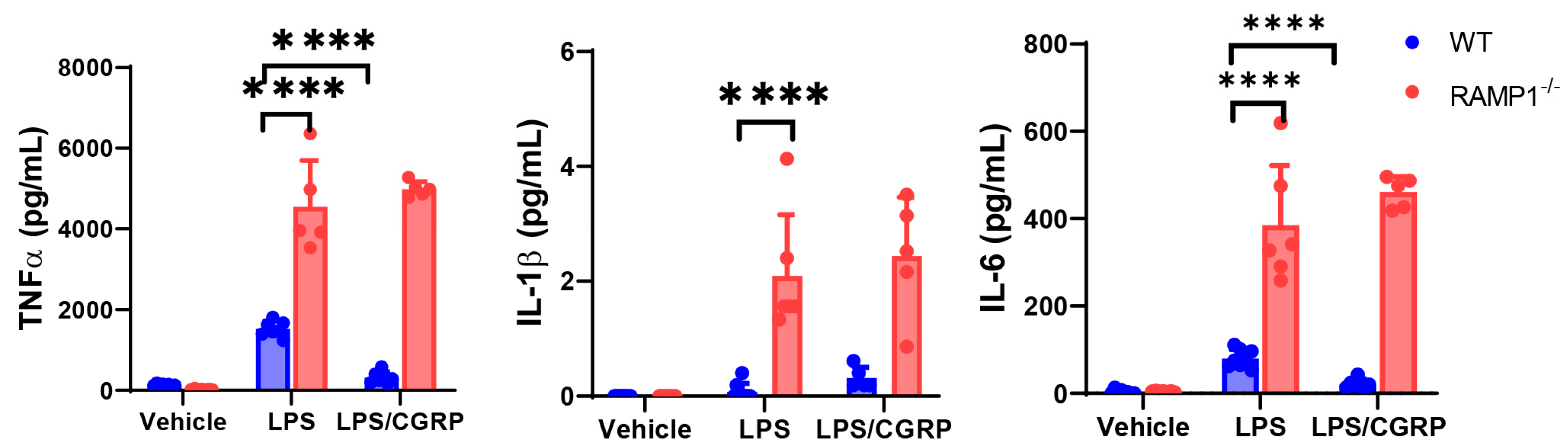
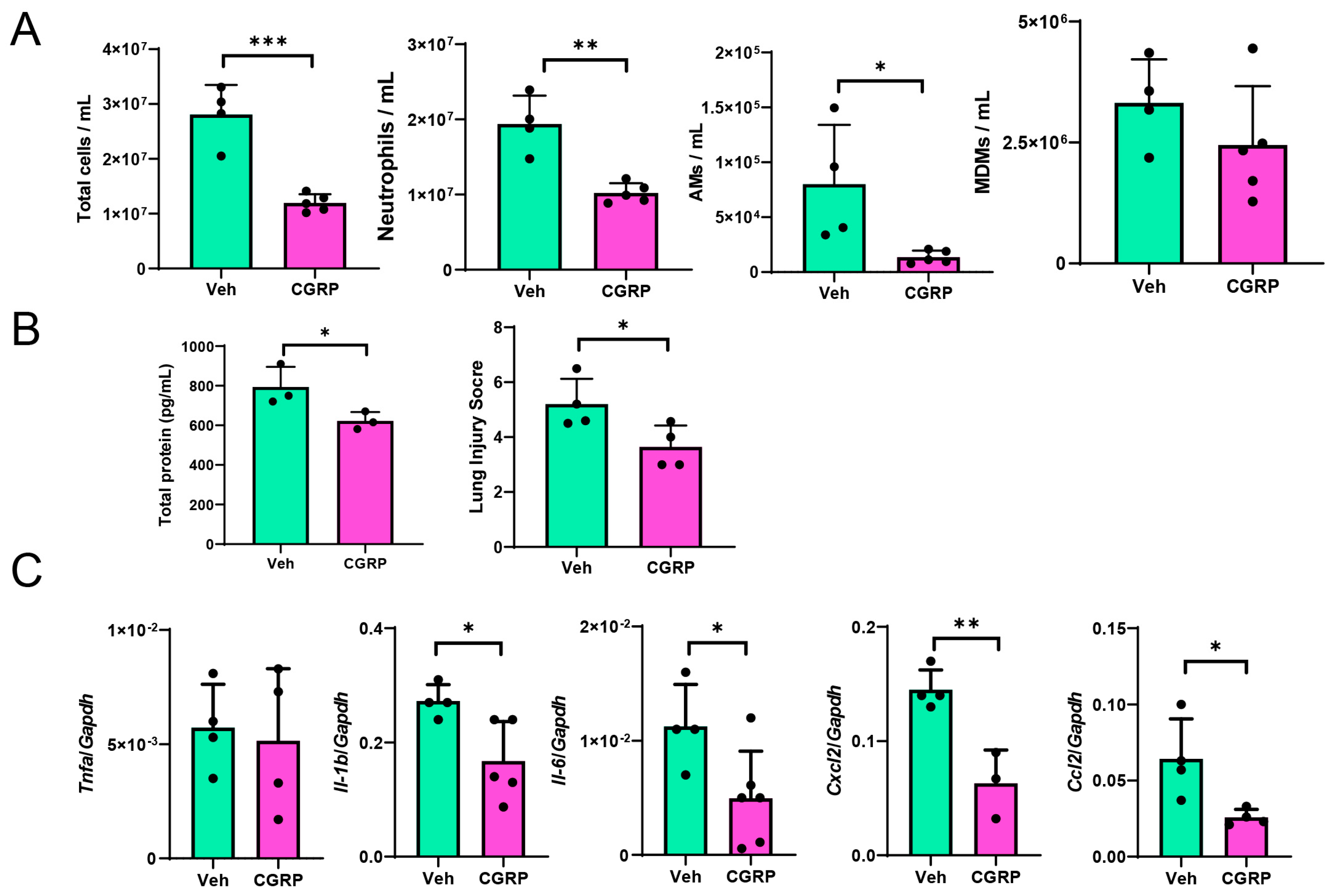
2.8. CGRP Treatment Mitigates LPS-Induced ALI
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Protocols
4.3. Bronchoalveolar Lavage (BAL)
4.4. Immune Cell Depletion
4.5. Flow Cytometry Analysis
4.6. Cell Sorting
4.7. Quantification of Cytokines and Chemokines
4.8. AM Culture
4.9. BM-Derived Macrophage Culture
4.10. Adoptive Transfer of AMs
4.11. Dissection of Dorsal Root Ganglion (DRG)
4.12. Histology and Immunohistochemical Analysis
4.13. Immunofluorescence
4.14. Quantitative Real-Time Reverse Transcription-PCR Analysis
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Lomas-Neira, J.; Chung, C.S.; Perl, M.; Gregory, S.; Biffl, W.; Ayala, A. Role of alveolar macrophage and migrating neutrophils in hemorrhage-induced priming for ALI subsequent to septic challenge. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L51–L58. [Google Scholar] [CrossRef]
- Hewitt, R.J.; Lloyd, C.M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Jean, E.E.; Good, O.; Rico, J.M.I.; Rossi, H.L.; Herbert, D.R. Neuroimmune regulatory networks of the airway mucosa in allergic inflammatory disease. J. Leukoc. Biol. 2022, 111, 209–221. [Google Scholar] [CrossRef]
- Verastegui, C.; Oliveira, A.P.; Fernandez-Vivero, J.; Romero, A.; de Castro, J.M. Calcitonin gene-related peptide immunoreactivity in adult mouse lung. Eur. J. Histochem. 1997, 41, 119–126. [Google Scholar] [PubMed]
- Hay, D.L.; Christopoulos, G.; Christopoulos, A.; Poyner, D.R.; Sexton, P.M. Pharmacological discrimination of calcitonin receptor: Receptor activity-modifying protein complexes. Mol. Pharmacol. 2005, 67, 1655–1665. [Google Scholar] [CrossRef]
- Majima, M.; Ito, Y.; Hosono, K.; Amano, H. CGRP/CGRP Receptor Antibodies: Potential Adverse Effects Due to Blockade of Neovascularization? Trends Pharmacol. Sci. 2019, 40, 11–21. [Google Scholar] [CrossRef]
- McLatchie, L.M.; Fraser, N.J.; Main, M.J.; Wise, A.; Brown, J.; Thompson, N.; Solari, R.; Lee, M.G.; Foord, S.M. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 1998, 393, 333–339. [Google Scholar] [CrossRef]
- Tsujikawa, K.; Yayama, K.; Hayashi, T.; Matsushita, H.; Yamaguchi, T.; Shigeno, T.; Ogitani, Y.; Hirayama, M.; Kato, T.; Fukada, S.; et al. Hypertension and dysregulated proinflammatory cytokine production in receptor activity-modifying protein 1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 16702–16707. [Google Scholar] [CrossRef]
- Inoue, T.; Ito, Y.; Nishizawa, N.; Eshima, K.; Kojo, K.; Otaka, F.; Betto, T.; Yamane, S.; Tsujikawa, K.; Koizumi, W.; et al. RAMP1 in Kupffer cells is a critical regulator in immune-mediated hepatitis. PLoS ONE 2018, 13, e0200432. [Google Scholar] [CrossRef]
- Kawashima-Takeda, N.; Ito, Y.; Nishizawa, N.; Kawashima, R.; Tanaka, K.; Tsujikawa, K.; Watanabe, M.; Majima, M. RAMP1 suppresses mucosal injury from dextran sodium sulfate-induced colitis in mice. J. Gastroenterol. Hepatol. 2017, 32, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.; Umans, B.D.; Li, L.; Wallrapp, A.; Bist, M.; Kirschbaum, T.; Wei, Y.; Zhou, Y.; Kuchroo, V.K.; Burkett, P.R.; et al. Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat. Med. 2018, 24, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xv, M.; Yang, W.C.; Wang, N.; Zhang, X.Z.; Li, W.Z. Exogenous alpha-calcitonin gene-related peptide attenuates lipopolysaccharide-induced acute lung injury in rats. Mol. Med. Rep. 2015, 12, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.-M.; Huangfu, C.-R.; Zheng, R.; Su, L.-N.; Wang, Y.-J.; Li, L. CGRP 8-37 enhances lipopolysaccharide-induced acute lung injury and regulating aquaporin 1 and 5 expressions in rats. J. Physiol. Biochem. 2016, 73, 381–386. [Google Scholar] [CrossRef]
- Ochoa-Callejero, L.; Garcia-Sanmartin, J.; Villoslada-Blanco, P.; Iniguez, M.; Perez-Matute, P.; Pujadas, E.; Fowkes, M.E.; Brody, R.; Oteo, J.A.; Martinez, A. Circulating Levels of Calcitonin Gene-Related Peptide Are Lower in COVID-19 Patients. J. Endocr. Soc. 2021, 5, bvaa199. [Google Scholar] [CrossRef]
- Rizzi, M.; Tonello, S.; Morani, F.; Rizzi, E.; Casciaro, G.F.; Matino, E.; Costanzo, M.; Zecca, E.; Croce, A.; Pedrinelli, A.; et al. CGRP Plasma Levels Correlate with the Clinical Evolution and Prognosis of Hospitalized Acute COVID-19 Patients. Viruses 2022, 14, 2123. [Google Scholar] [CrossRef]
- Hou, F.; Xiao, K.; Tang, L.; Xie, L. Diversity of Macrophages in Lung Homeostasis and Diseases. Front. Immunol. 2021, 12, 753940. [Google Scholar] [CrossRef]
- Cohen, S.B.; Gern, B.H.; Delahaye, J.L.; Adams, K.N.; Plumlee, C.R.; Winkler, J.K.; Sherman, D.R.; Gerner, M.Y.; Urdahl, K.B. Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe 2018, 24, 439–446.e4. [Google Scholar] [CrossRef]
- Dey, A.; Allen, J.; Hankey-Giblin, P.A. Ontogeny and polarization of macrophages in inflammation: Blood monocytes versus tissue macrophages. Front. Immunol. 2014, 5, 683. [Google Scholar] [CrossRef]
- Mould, K.J.; Barthel, L.; Mohning, M.P.; Thomas, S.M.; McCubbrey, A.L.; Danhorn, T.; Leach, S.M.; Fingerlin, T.E.; O’Connor, B.P.; Reisz, J.A.; et al. Cell Origin Dictates Programming of Resident versus Recruited Macrophages during Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2017, 57, 294–306. [Google Scholar] [CrossRef]
- Bain, C.C.; MacDonald, A.S. The impact of the lung environment on macrophage development, activation and function: Diversity in the face of adversity. Mucosal Immunol. 2022, 15, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Keith, I.M.; Pelto-Huikko, M.; Schalling, M.; Hokfelt, T. Calcitonin gene-related peptide and its mRNA in pulmonary neuroendocrine cells and ganglia. Histochemistry 1991, 96, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Barr, J.; Jaquish, A.; Xu, J.; Verheyden, J.M.; Sun, X. Identification of lung innervating sensory neurons and their target specificity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L50–L63. [Google Scholar] [CrossRef]
- Puttur, F.; Gregory, L.G.; Lloyd, C.M. Airway macrophages as the guardians of tissue repair in the lung. Immunol. Cell Biol. 2019, 97, 246–257. [Google Scholar] [CrossRef]
- Coleridge, J.C.; Coleridge, H.M. Afferent vagal C fibre innervation of the lungs and airways and its functional significance. Rev. Physiol. Biochem. Pharmacol. 1984, 99, 1–110. [Google Scholar] [CrossRef] [PubMed]
- Le, D.D.; Rochlitzer, S.; Fischer, A.; Heck, S.; Tschernig, T.; Sester, M.; Bals, R.; Welte, T.; Braun, A.; Dinh, Q.T. Allergic airway inflammation induces the migration of dendritic cells into airway sensory ganglia. Respir. Res. 2014, 15, 73. [Google Scholar] [CrossRef]
- Wallrapp, A.; Burkett, P.R.; Riesenfeld, S.J.; Kim, S.J.; Christian, E.; Abdulnour, R.E.; Thakore, P.I.; Schnell, A.; Lambden, C.; Herbst, R.H.; et al. Calcitonin Gene-Related Peptide Negatively Regulates Alarmin-Driven Type 2 Innate Lymphoid Cell Responses. Immunity 2019, 51, 709–723.e6. [Google Scholar] [CrossRef]
- Li, M.; Wetzel-Strong, S.E.; Hua, X.; Tilley, S.L.; Oswald, E.; Krummel, M.F.; Caron, K.M. Deficiency of RAMP1 attenuates antigen-induced airway hyperresponsiveness in mice. PLoS ONE 2014, 9, e102356. [Google Scholar] [CrossRef]
- Russo, A.F.; Hay, D.L. CGRP physiology, pharmacology, and therapeutic targets: Migraine and beyond. Physiol. Rev. 2023, 103, 1565–1644. [Google Scholar] [CrossRef]
- Joshi, N.; Walter, J.M.; Misharin, A.V. Alveolar Macrophages. Cell Immunol. 2018, 330, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Leemans, J.C.; Florquin, S.; Branger, J.; Maris, N.A.; Pater, J.; van Rooijen, N.; van der Poll, T. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 2003, 167, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Broug-Holub, E.; Toews, G.B.; van Iwaarden, J.F.; Strieter, R.M.; Kunkel, S.L.; Paine, R., 3rd; Standiford, T.J. Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: Elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect. Immun. 1997, 65, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Wang, H.; Zheng, K.; Yang, W.; Xiao, K.; Rong, Z.; Xiao, J.; Li, J.; Cheng, B.; Tang, L.; et al. Distinct Transcriptional and Functional Differences of Lung Resident and Monocyte-Derived Alveolar Macrophages During the Recovery Period of Acute Lung Injury. Immune Netw. 2023, 23, e24. [Google Scholar] [CrossRef]
- Aegerter, H.; Lambrecht, B.N.; Jakubzick, C.V. Biology of lung macrophages in health and disease. Immunity 2022, 55, 1564–1580. [Google Scholar] [CrossRef]
- Mould, K.J.; Jackson, N.D.; Henson, P.M.; Seibold, M.; Janssen, W.J. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight 2019, 4, e126556. [Google Scholar] [CrossRef]
- Aegerter, H.; Kulikauskaite, J.; Crotta, S.; Patel, H.; Kelly, G.; Hessel, E.M.; Mack, M.; Beinke, S.; Wack, A. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat. Immunol. 2020, 21, 145–157. [Google Scholar] [CrossRef]
- Han, W.; Tanjore, H.; Liu, Y.; Hunt, R.P.; Gutor, S.S.; Serezani, A.P.M.; Blackwell, T.S. Identification and Characterization of Alveolar and Recruited Lung Macrophages during Acute Lung Inflammation. J. Immunol. 2023, 210, 1827–1836. [Google Scholar] [CrossRef]
- Chen, L.; Sun, R.; Lei, C.; Xu, Z.; Song, Y.; Deng, Z. Alcohol-mediated susceptibility to lung fibrosis is associated with group 2 innate lymphoid cells in mice. Front. Immunol. 2023, 14, 1178498. [Google Scholar] [CrossRef]
- Jusek, G.; Reim, D.; Tsujikawa, K.; Holzmann, B. Deficiency of the CGRP receptor component RAMP1 attenuates immunosuppression during the early phase of septic peritonitis. Immunobiology 2012, 217, 761–767. [Google Scholar] [CrossRef]
- Steinberg, K.P.; Milberg, J.A.; Martin, T.R.; Maunder, R.J.; Cockrill, B.A.; Hudson, L.D. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1994, 150, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Potey, P.M.; Rossi, A.G.; Lucas, C.D.; Dorward, D.A. Neutrophils in the initiation and resolution of acute pulmonary inflammation: Understanding biological function and therapeutic potential. J. Pathol. 2019, 247, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Rebetz, J.; Semple, J.W.; Kapur, R. The Pathogenic Involvement of Neutrophils in Acute Respiratory Distress Syndrome and Transfusion-Related Acute Lung Injury. Transfus. Med. Hemother. 2018, 45, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, Y.; Suzuki, T.; Kuniyoshi, K.; Inamo, J.; Yamaguchi, K.; Komuro, M.; Watanabe, J.; Edamoto, M.; Li, S.; Kouno, T.; et al. Expression of the readthrough transcript CiDRE in alveolar macrophages boosts SARS-CoV-2 susceptibility and promotes COVID-19 severity. Immunity 2023, 56, 1939–1954.e12. [Google Scholar] [CrossRef] [PubMed]
- Hosono, K.; Yamashita, A.; Tanabe, M.; Ito, Y.; Majima, M.; Tsujikawa, K.; Amano, H. Deletion of RAMP1 Signaling Enhances Diet-induced Obesity and Fat Absorption via Intestinal Lacteals in Mice. In Vivo 2024, 38, 160–173. [Google Scholar] [CrossRef]
- Su, X.; Wang, L.; Song, Y.; Bai, C. Inhibition of inflammatory responses by ambroxol, a mucolytic agent, in a murine model of acute lung injury induced by lipopolysaccharide. Intensive Care Med. 2004, 30, 133–140. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamashita, A.; Ito, Y.; Osada, M.; Matsuda, H.; Hosono, K.; Tsujikawa, K.; Okamoto, H.; Amano, H. RAMP1 Signaling Mitigates Acute Lung Injury by Distinctively Regulating Alveolar and Monocyte-Derived Macrophages. Int. J. Mol. Sci. 2024, 25, 10107. https://doi.org/10.3390/ijms251810107
Yamashita A, Ito Y, Osada M, Matsuda H, Hosono K, Tsujikawa K, Okamoto H, Amano H. RAMP1 Signaling Mitigates Acute Lung Injury by Distinctively Regulating Alveolar and Monocyte-Derived Macrophages. International Journal of Molecular Sciences. 2024; 25(18):10107. https://doi.org/10.3390/ijms251810107
Chicago/Turabian StyleYamashita, Atsushi, Yoshiya Ito, Mayuko Osada, Hiromi Matsuda, Kanako Hosono, Kazutake Tsujikawa, Hirotsugu Okamoto, and Hideki Amano. 2024. "RAMP1 Signaling Mitigates Acute Lung Injury by Distinctively Regulating Alveolar and Monocyte-Derived Macrophages" International Journal of Molecular Sciences 25, no. 18: 10107. https://doi.org/10.3390/ijms251810107