

Reliability of AlphaFold2 Models in Virtual Drug Screening: A Focus on Selected Class A GPCRs

Abstract
:1. Introduction
2. Results
2.1. Quality of Class A GPCR AlphaFold2 Models
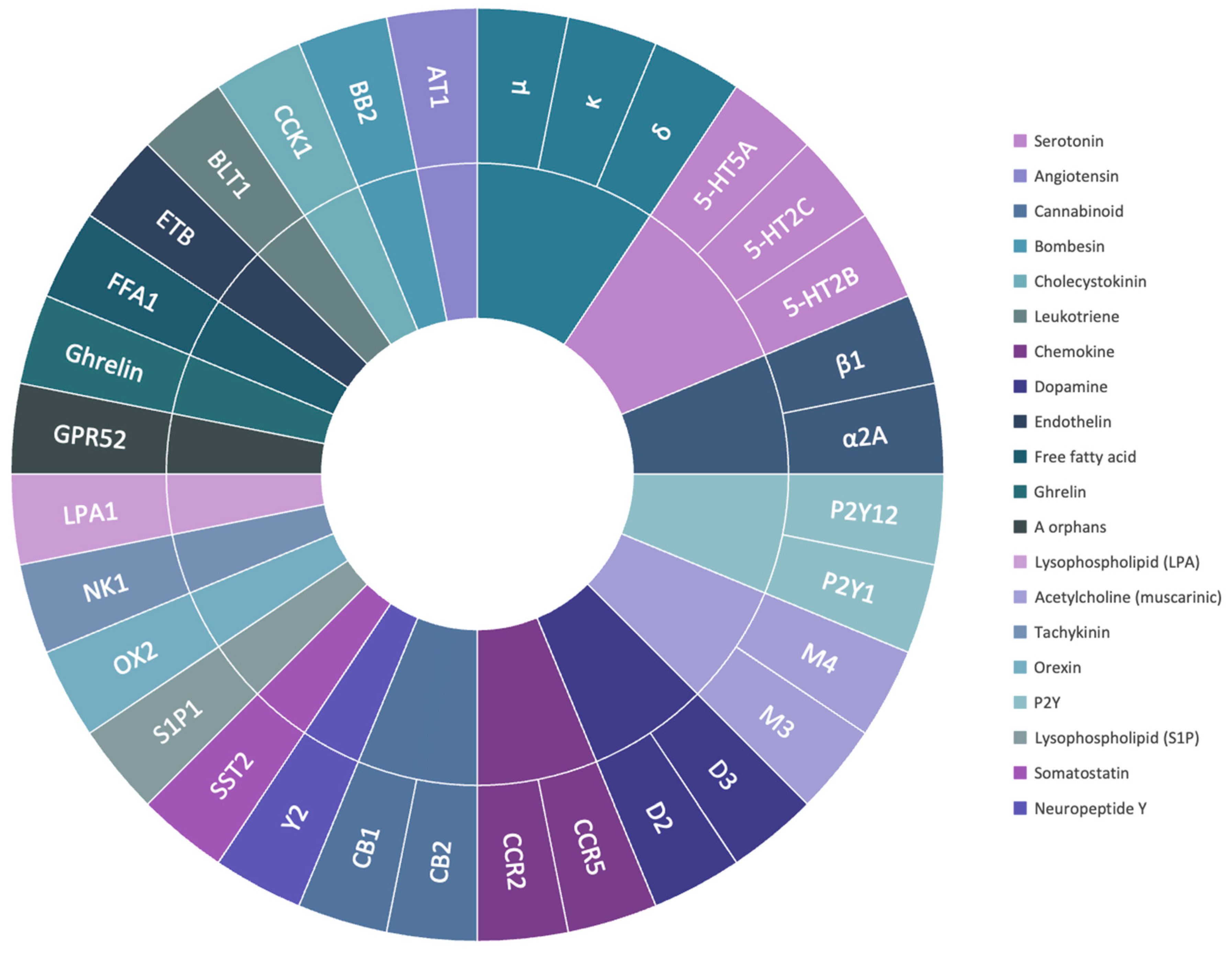
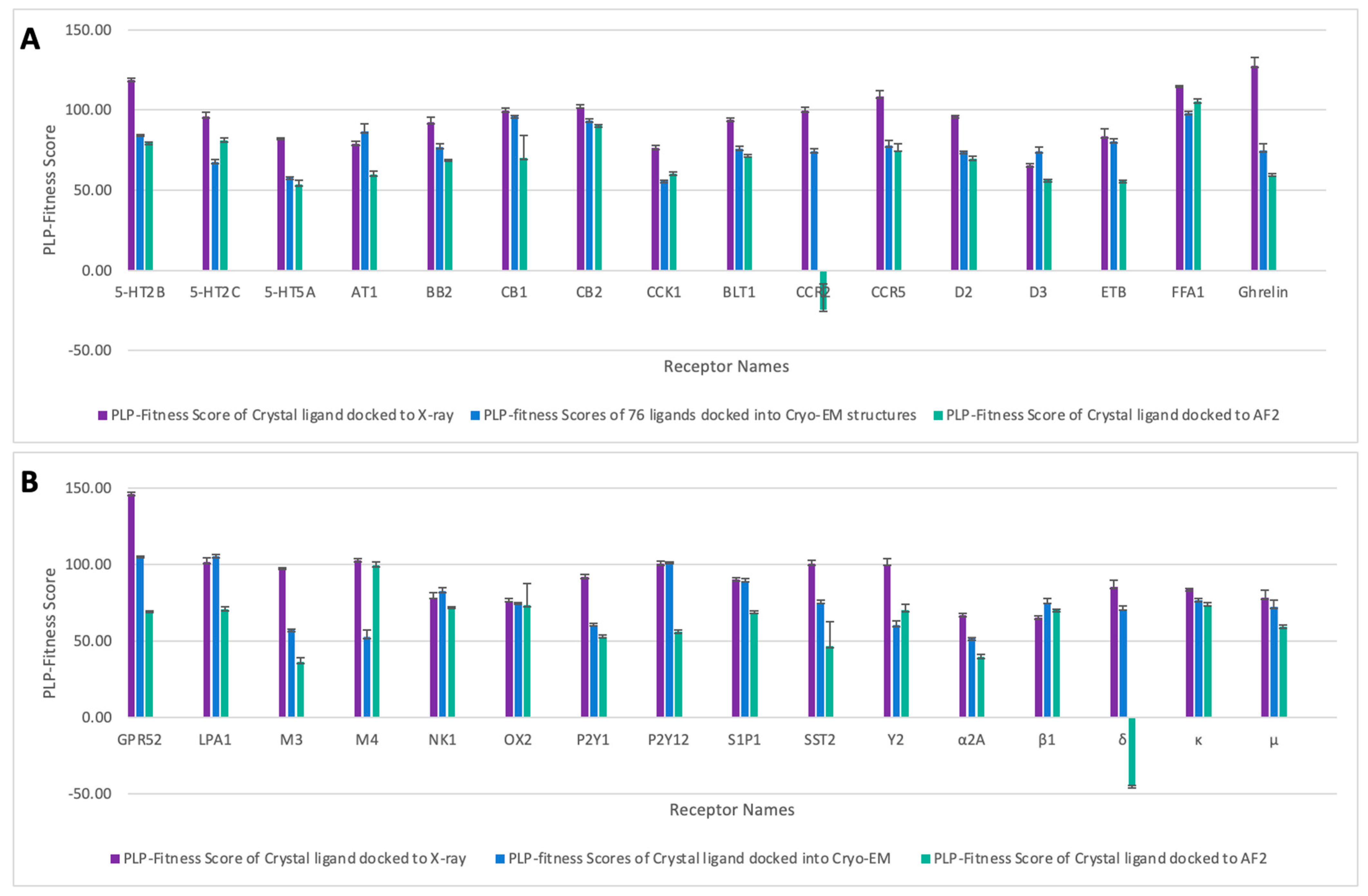
2.2. Molecular Docking Scores of the Selected Class A GPCRs
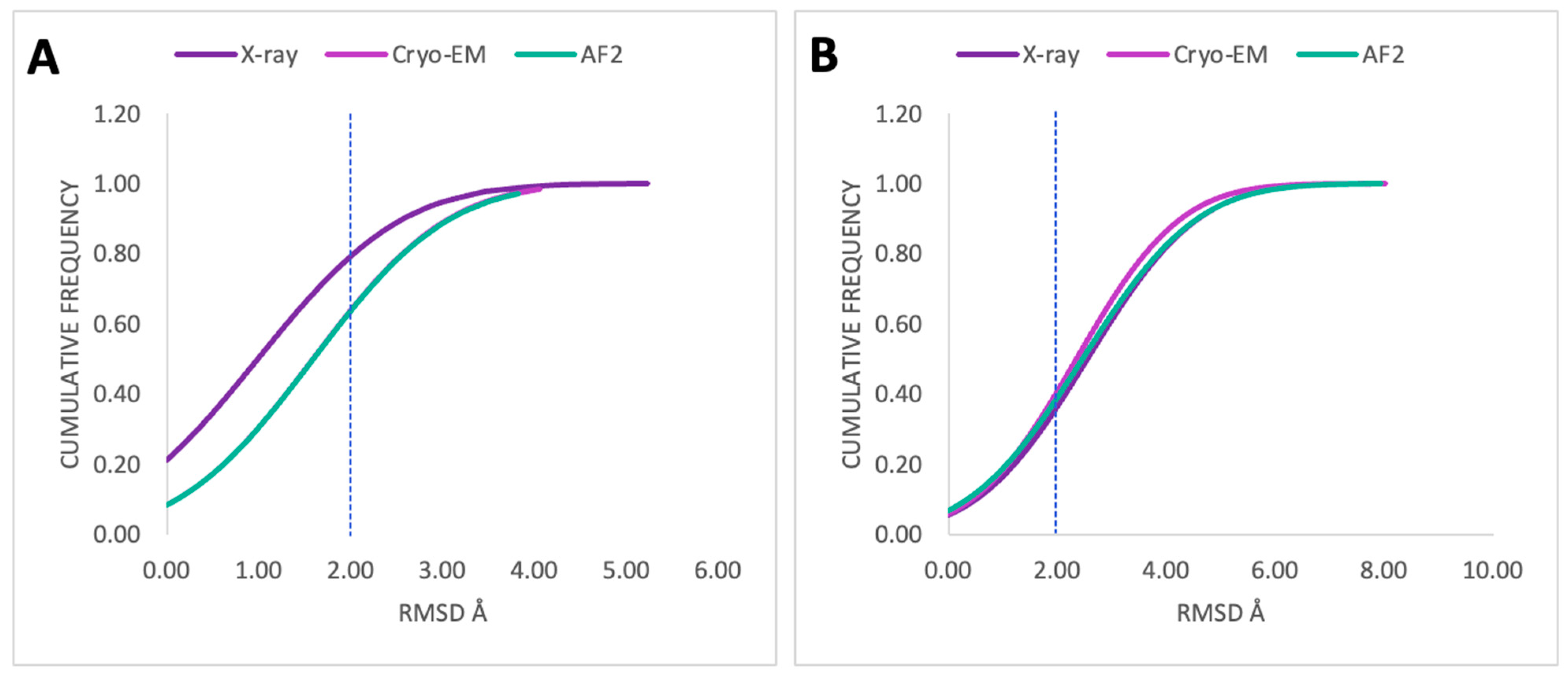
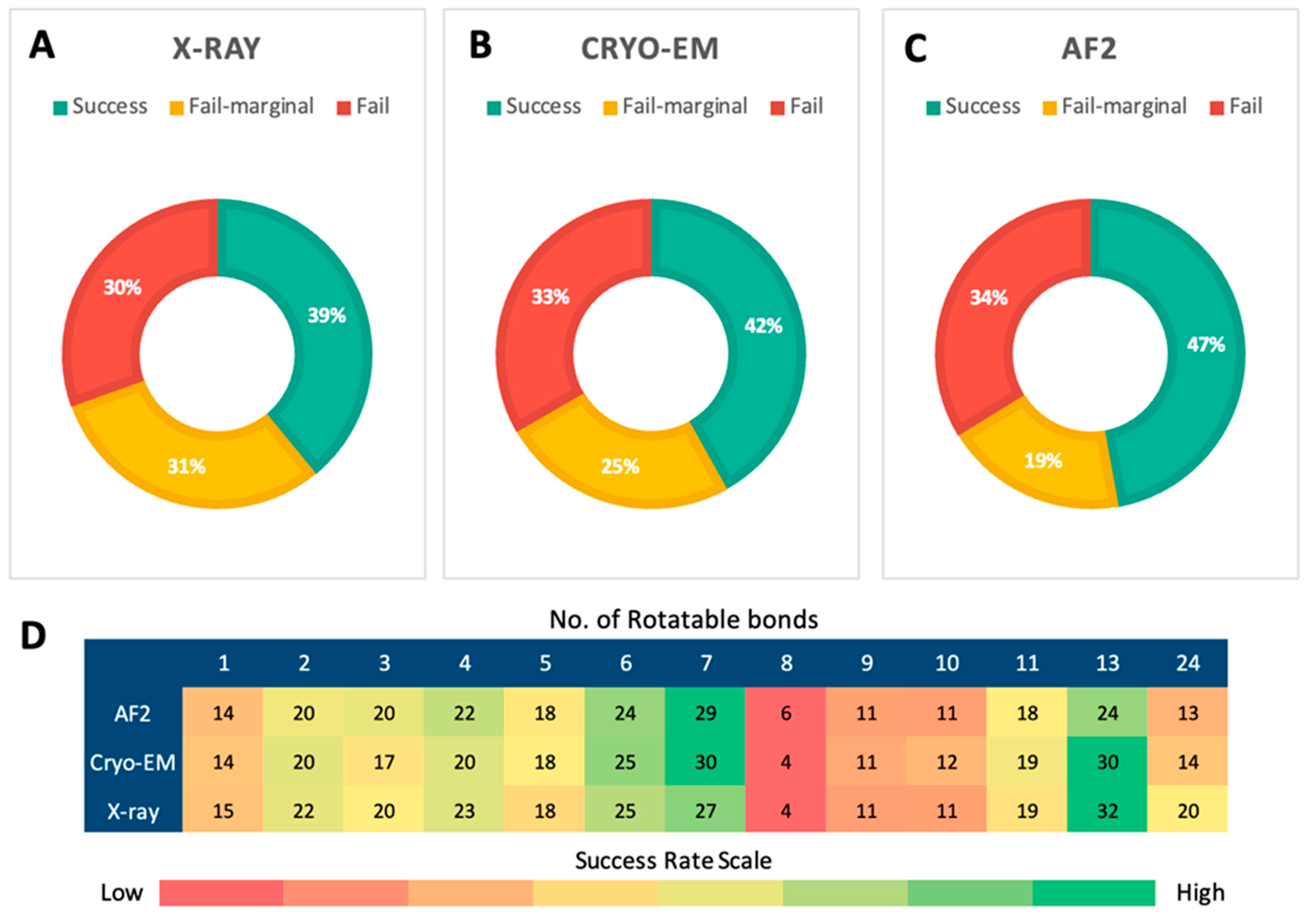
2.3. Posing Accuracy of Docked Ligands
2.4. Evaluation of Screening Power
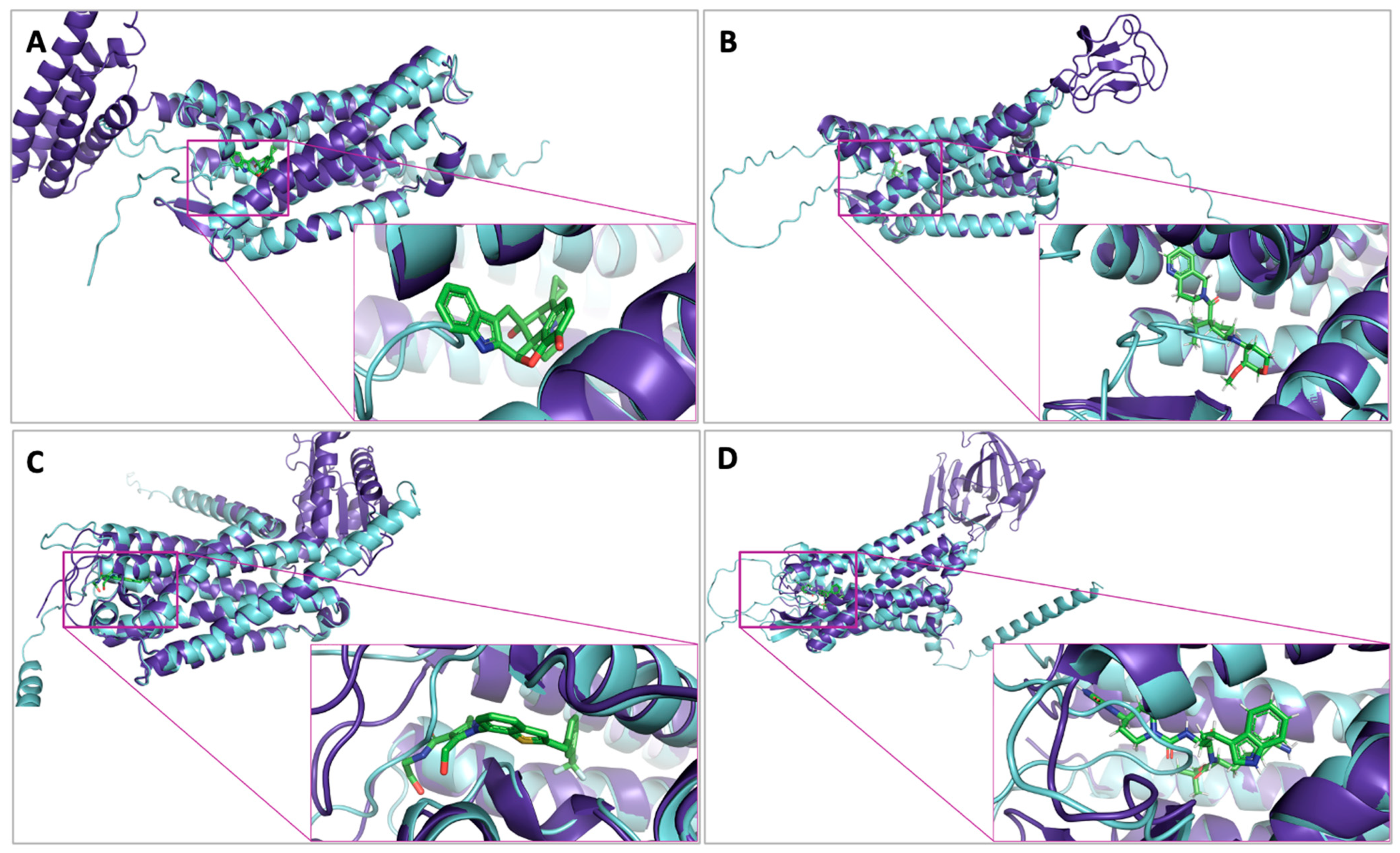
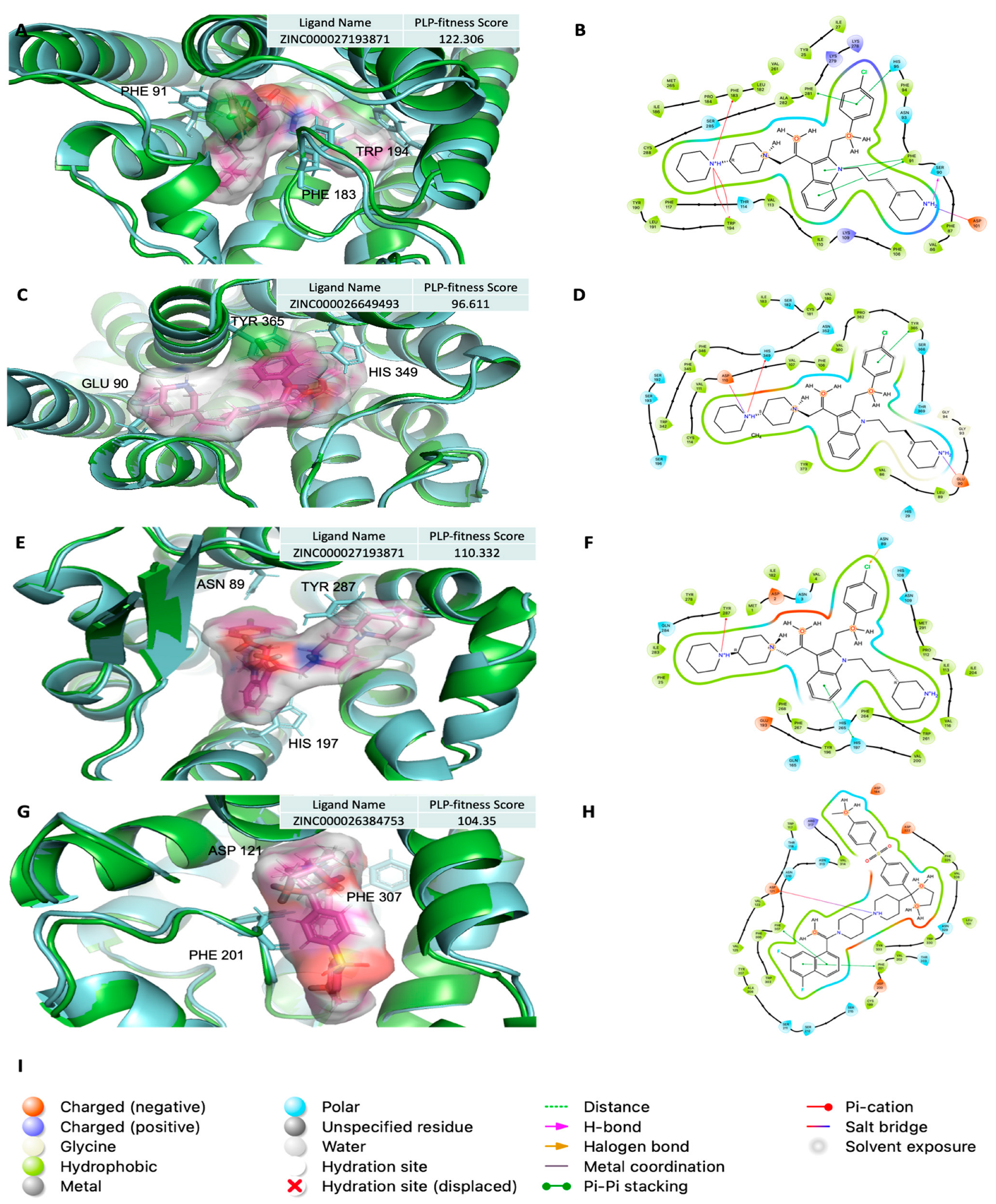
2.5. Analysis of Ligand Competitive Inhibition
3. Discussion
4. Materials and Methods
4.1. Construction of Ligand Library
4.2. Preparation of Receptors
4.3. Assessment of AlphaFold2 Structures
4.4. Overview of Molecular Docking
4.5. Assessment Methods
4.5.1. GOLD’s ChemPLP Scoring Function
4.5.2. Root Mean Square Deviation (RMSD)
4.5.3. Enrichment Factor (EF)
4.6. Ligand Competitive Inhibition Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Varadi, M.; Velankar, S. The impact of AlphaFold Protein Structure Database on the fields of life sciences. Proteomics 2023, 23, 2200128. [Google Scholar] [CrossRef] [PubMed]
- AlQuraishi, M. Protein-structure prediction revolutionized. Nature 2021, 596, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Hekkelman, M.L.; de Vries, I.; Joosten, R.P.; Perrakis, A. AlphaFill: Enriching AlphaFold models with ligands and cofactors. Nat. Methods 2023, 20, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2023, 52, D368–D375. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins Struct. Funct. Bioinform. 2019, 87, 1141–1148. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Bouatta, N.; Sorger, P.; AlQuraishi, M. Protein structure prediction by AlphaFold2: Are attention and symmetries all you need? Acta Crystallogr. Sect. Struct. Biol. 2021, 77, 982–991. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.O.; He, Y. Benchmarking the Accuracy of AlphaFold 2 in Loop Structure Prediction. Biomolecules 2022, 12, 985. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.C.; Liebschner, D.; Croll, T.I.; Williams, C.J.; McCoy, A.J.; Poon, B.K.; Afonine, P.V.; Oeffner, R.D.; Richardson, J.S.; Read, R.J.; et al. AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nat. Methods 2024, 21, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Michino, M.; Abola, E.; Brooks, C.L.; Dixon, J.S.; Moult, J.; Stevens, R.C. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat. Rev. Drug Discov. 2009, 8, 455–463. [Google Scholar] [CrossRef]
- Xu, H.E.; Xiao, R. A new era for GPCR research: Structures, biology and drug discovery. Acta Pharmacol. Sin. 2012, 33, 289–290. [Google Scholar] [CrossRef]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G protein-coupled receptors: Structure- and function-based drug discovery. Signal Transduct. Target. Ther. 2021, 6, 1–27. [Google Scholar] [CrossRef]
- Hu, G.-M.; Mai, T.-L.; Chen, C.-M. Visualizing the GPCR Network: Classification and Evolution. Sci. Rep. 2017, 7, 15495. [Google Scholar] [CrossRef]
- Krumm, B.; Roth, B.L. A Structural Understanding of Class B GPCR Selectivity and Activation Revealed. Structure 2020, 28, 277–279. [Google Scholar] [CrossRef]
- Laeremans, T.; Sands, Z.A.; Claes, P.; De Blieck, A.; De Cesco, S.; Triest, S.; Busch, A.; Felix, D.; Kumar, A.; Jaakola, V.-P.; et al. Accelerating GPCR Drug Discovery With Conformation-Stabilizing VHHs. Front. Mol. Biosci. 2022, 9, 863099. [Google Scholar] [CrossRef] [PubMed]
- He, X.; You, C.; Jiang, H.; Jiang, Y.; Xu, H.E.; Cheng, X. AlphaFold2 versus experimental structures: Evaluation on G protein-coupled receptors. Acta Pharmacol. Sin. 2023, 44, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Scardino, V.; Di Filippo, J.I.; Cavasotto, C.N. How good are AlphaFold models for docking-based virtual screening? iScience 2023, 26, 105920. [Google Scholar] [CrossRef] [PubMed]
- Karelina, M.; Noh, J.J.; Dror, R.O. How accurately can one predict drug binding modes using AlphaFold models? eLife 2023, 12, RP89386. [Google Scholar] [CrossRef] [PubMed]
- Harmalkar, A.; Lyskov, S.; Gray, J.J. Reliable protein-protein docking with AlphaFold, Rosetta, and replica-exchange. eLife 2024, 13. [Google Scholar] [CrossRef]
- Holcomb, M.; Chang, Y.-T.; Goodsell, D.S.; Forli, S. Evaluation of AlphaFold2 structures as docking targets. Protein Sci. Publ. Protein Soc. 2023, 32, e4530. [Google Scholar] [CrossRef]
- Zhang, Y.; Vass, M.; Shi, D.; Abualrous, E.; Chambers, J.M.; Chopra, N.; Higgs, C.; Kasavajhala, K.; Li, H.; Nandekar, P.; et al. Benchmarking Refined and Unrefined AlphaFold2 Structures for Hit Discovery. J. Chem. Inf. Model. 2023, 63, 1656–1667. [Google Scholar] [CrossRef]
- Wong, F.; Krishnan, A.; Zheng, E.J.; Stärk, H.; Manson, A.L.; Earl, A.M.; Jaakkola, T.; Collins, J.J. Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol. 2022, 18, e11081. [Google Scholar] [CrossRef]
- Marcu, Ş.-B.; Tăbîrcă, S.; Tangney, M. An Overview of Alphafold’s Breakthrough. Front. Artif. Intell. 2022, 5, 875587. [Google Scholar] [CrossRef]
- Bertoline, L.M.F.; Lima, A.N.; Krieger, J.E.; Teixeira, S.K. Before and after AlphaFold2: An overview of protein structure prediction. Front. Bioinform. 2023, 3, 1120370. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. Multi-state modeling of G-protein coupled receptors at experimental accuracy. Proteins Struct. Funct. Bioinform. 2022, 90, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Aided Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. PCCP 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Chaput, L.; Selwa, E.; Elisée, E.; Iorga, B.I. Blinded evaluation of cathepsin S inhibitors from the D3RGC3 dataset using molecular docking and free energy calculations. J. Comput. Aided Mol. Des. 2019, 33, 93–103. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking1. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef]
- Anighoro, A.; De La Vega De León, A.; Bajorath, J. Predicting bioactive conformations and binding modes of macrocycles. J. Comput. Aided Mol. Des. 2016, 30, 841–849. [Google Scholar] [CrossRef]
- Sasmal, S.; Gill, S.C.; Lim, N.M.; Mobley, D.L. Sampling Conformational Changes of Bound Ligands Using Nonequilibrium Candidate Monte Carlo and Molecular Dynamics. J. Chem. Theory Comput. 2020, 16, 1854–1865. [Google Scholar] [CrossRef]
- Fischer, M.; Coleman, R.G.; Fraser, J.S.; Shoichet, B.K. The incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem. 2014, 6, 575–583. [Google Scholar] [CrossRef]
- Ren, F.; Ding, X.; Zheng, M.; Korzinkin, M.; Cai, X.; Zhu, W.; Mantsyzov, A.; Aliper, A.; Aladinskiy, V.; Cao, Z.; et al. AlphaFold accelerates artificial intelligence powered drug discovery: Efficient discovery of a novel CDK20 small molecule inhibitor. Chem. Sci. 2023, 14, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Baselious, F.; Hilscher, S.; Robaa, D.; Barinka, C.; Schutkowski, M.; Sippl, W. Comparative Structure-Based Virtual Screening Utilizing Optimized AlphaFold Model Identifies Selective HDAC11 Inhibitor. Int. J. Mol. Sci. 2024, 25, 1358. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Zhang, M.; Liu, Y.; Jang, H. AlphaFold, allosteric, and orthosteric drug discovery: Ways forward. Drug Discov. Today 2023, 28, 103551. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Aucar, M.G.; Adler, N.S. Computational chemistry in drug lead discovery and design. Int. J. Quantum Chem. 2019, 119, e25678. [Google Scholar] [CrossRef]
- Stampelou, M.; Ladds, G.; Kolocouris, A. Computational Workflow for Refining AlphaFold Models in Drug Design Using Kinetic and Thermodynamic Binding Calculations: A Case Study for the Unresolved Inactive Human Adenosine A3 Receptor. J. Phys. Chem. B 2024, 128, 914–936. [Google Scholar] [CrossRef]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- ChEMBL Database in 2023: A Drug Discovery Platform Spanning Multiple Bioactivity Data Types and Time Periods|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/52/D1/D1180/7337608 (accessed on 25 February 2024).
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt. The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Hymavati; Kumar, V.; Elizabeth Sobhia, M. Implication of Crystal Water Molecules in Inhibitor Binding at ALR2 Active Site. Comput. Math. Methods Med. 2012, 2012, 541594. [Google Scholar] [CrossRef]
- Speers, A.E.; Cravatt, B.F. Ligands in crystal structures that aid in functional characterization. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; He, H.; Wang, J.; Wang, J.; Chang, C.A. Uncovering Water Effects in Protein–Ligand Recognition: Importance in the Second Hydration Shell and Binding Kinetics. Phys. Chem. Chem. Phys. PCCP 2023, 25, 2098–2109. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Alogheli, H.; Olanders, G.; Schaal, W.; Brandt, P.; Karlén, A. Docking of Macrocycles: Comparing Rigid and Flexible Docking in Glide. J. Chem. Inf. Model. 2017, 57, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Nicholls, A. Conformer Generation with OMEGA: Learning from the Data Set and the Analysis of Failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Scardino, V.; Bollini, M.; Cavasotto, C.N. Combination of pose and rank consensus in docking-based virtual screening: The best of both worlds. RSC Adv. 2021, 11, 35383–35391. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | pLDDT (Global) Score | Backbone RMSD (Å) | Binding Site RMSD (Å) | MolProbity Score | Ramachandran Favored (%) | QMEAN Z-Score | QMEANDisCo Global |
|---|---|---|---|---|---|---|---|
| 5-HT2B | 71.74 | 0.93 | 0.45 | 2.09 | 88.10 | −6.49 | 0.59 ± 0.05 |
| 5-HT2C | 73.53 | 0.52 | 0.44 | 1.78 | 87.28 | −6.15 | 0.58 ± 0.05 |
| 5-HT5A | 78.89 | 1.11 | 0.66 | 2.09 | 87.61 | −4.39 | 0.64 ± 0.05 |
| AT1 | 82.04 | 1.07 | 0.48 | 1.55 | 91.04 | −3.33 | 0.67 ± 0.05 |
| BB2 | 78.92 | 1.15 | 0.90 | 1.97 | 90.05 | −5.67 | 0.61 ± 0.05 |
| CB1 | 71.66 | 1.97 | 1.17 | 1.67 | 90.21 | −5.08 | 0.59 ± 0.05 |
| CB2 | 82.48 | 0.87 | 0.38 | 1.69 | 93.30 | −4.08 | 0.70 ± 0.05 |
| CCK1 | 78.54 | 0.81 | 0.75 | 1.89 | 89.20 | −4.96 | 0.61 ± 0.05 |
| BLT1 | 82.53 | 0.88 | 0.47 | 1.77 | 91.14 | −4.91 | 0.68 ± 0.05 |
| CCR2 | 78.28 | 0.63 | 0.50 | 1.66 | 86.83 | −5.72 | 0.64 ± 0.05 |
| CCR5 | 85.29 | 0.77 | 0.69 | 1.42 | 92.57 | −4.46 | 0.70 ± 0.05 |
| D2 | 72.41 | 0.81 | 0.52 | 2.18 | 87.76 | −5.07 | 0.54 ± 0.05 |
| D3 | 75.64 | 0.50 | 0.30 | 1.80 | 91.96 | −4.52 | 0.61 ± 0.05 |
| ETB | 75.52 | 1.56 | 0.54 | 1.90 | 90.45 | −5.27 | 0.66 ± 0.05 |
| FFA1 | 89.45 | 0.55 | 0.37 | 1.44 | 94.97 | −3.29 | 0.77 ± 0.05 |
| Ghrelin | 81.36 | 1.54 | 0.59 | 1.41 | 95.05 | −2.12 | 0.69 ± 0.05 |
| GPR52 | 81.82 | 0.65 | 0.54 | 1.38 | 93.59 | −3.74 | 0.65 ± 0.05 |
| LPA1 | 84.50 | 0.37 | 0.27 | 1.69 | 90.33 | −4.97 | 0.70 ± 0.05 |
| M3 | 67.64 | 1.12 | 0.95 | 2.29 | 82.11 | −7.07 | 0.51 ± 0.06 |
| M4 | 76.23 | 0.42 | 0.24 | 1.77 | 88.68 | −6.16 | 0.54 ± 0.05 |
| NK1 | 78.25 | 0.46 | 0.31 | 1.74 | 90.62 | −4.65 | 0.66 ± 0.05 |
| OX2 | 78.27 | 0.53 | 0.27 | 1.72 | 92.76 | −3.57 | 0.65 ± 0.06 |
| P2Y1 | 86.18 | 0.64 | 0.79 | 1.34 | 91.37 | −4.30 | 0.72 ± 0.05 |
| P2Y12 | 85.04 | 0.79 | 0.45 | 1.84 | 91.47 | −4.13 | 0.70 ± 0.05 |
| S1P1 | 81.67 | 0.48 | 0.31 | 1.69 | 91.05 | −4.90 | 0.64 ± 0.05 |
| SST2 | 81.56 | 0.91 | 0.83 | 1.98 | 91.55 | −3.98 | 0.67 ± 0.05 |
| Y2 | 82.42 | 0.78 | 0.77 | 1.66 | 91.29 | −4.17 | 0.66 ± 0.06 |
| α2A | 72.11 | 0.85 | 0.49 | 1.61 | 84.45 | −6.13 | 0.53 ± 0.06 |
| β1 | 74.01 | 0.59 | 0.28 | 1.56 | 89.40 | −5.96 | 0.57 ± 0.05 |
| δ | 80.40 | 0.45 | 0.24 | 1.30 | 94.05 | −2.83 | 0.69 ± 0.05 |
| κ | 80.07 | 0.66 | 0.43 | 1.51 | 92.33 | −3.88 | 0.64 ± 0.05 |
| μ | 77.67 | 0.96 | 0.58 | 1.73 | 90.40 | −3.99 | 0.64 ± 0.06 |
| Receptor | X-ray Structures | Cryo-EM Structures | AlphaFold2 Models | ||||||
|---|---|---|---|---|---|---|---|---|---|
| EF | HR% | Correctly Classified Ligands% | EF | HR% | Correctly Classified Ligands% | EF | HR% | Correctly Classified Ligands% | |
| 5-HT2B | 2.53 | 33.33 | 13.16 | 3.28 | 43.33 | 17.11 | 2.02 | 26.67 | 10.53 |
| 5-HT2C | 2.27 | 30.00 | 11.84 | 2.53 | 33.33 | 13.16 | 2.02 | 26.67 | 10.53 |
| 5-HT5A | 1.52 | 20.00 | 7.89 | 1.26 | 16.67 | 6.58 | 1.26 | 16.67 | 6.58 |
| AT1 | 2.78 | 36.67 | 14.47 | 2.02 | 26.67 | 10.53 | 1.77 | 23.33 | 9.21 |
| BB2 | 1.77 | 23.33 | 9.21 | 4.04 | 53.33 | 21.05 | 2.78 | 36.67 | 14.47 |
| CB1 | 2.78 | 36.67 | 14.47 | 3.54 | 46.67 | 18.42 | 0.76 | 10.00 | 3.95 |
| CB2 | 2.27 | 30.00 | 11.84 | 2.53 | 33.33 | 13.16 | 1.01 | 13.33 | 5.26 |
| CCK1 | 2.27 | 30.00 | 11.84 | 2.53 | 33.33 | 13.16 | 2.78 | 36.67 | 14.47 |
| BLT1 | 3.03 | 40.00 | 15.79 | 1.77 | 23.33 | 9.21 | 2.27 | 30.00 | 11.84 |
| CCR2 | 2.53 | 33.33 | 13.16 | 2.53 | 33.33 | 13.16 | 0.00 | 0.00 | 0.00 |
| CCR5 | 2.53 | 33.33 | 13.16 | 2.02 | 26.67 | 10.53 | 2.53 | 33.33 | 13.16 |
| D2 | 2.27 | 30.00 | 11.84 | 3.79 | 50.00 | 19.74 | 2.02 | 26.67 | 10.53 |
| D3 | 1.01 | 13.33 | 5.26 | 1.26 | 16.67 | 6.58 | 1.26 | 16.67 | 6.58 |
| ETB | 1.52 | 20.00 | 7.89 | 3.54 | 46.67 | 18.42 | 1.26 | 16.67 | 6.58 |
| FFA1 | 2.78 | 36.67 | 14.47 | 2.78 | 36.67 | 14.47 | 1.26 | 16.67 | 6.58 |
| Ghrelin | 2.02 | 26.67 | 10.53 | 2.27 | 30.00 | 11.84 | 3.54 | 46.67 | 18.42 |
| GPR52 | 1.77 | 23.33 | 9.21 | 1.77 | 23.33 | 9.21 | 2.02 | 26.67 | 10.53 |
| LPA1 | 1.77 | 23.33 | 9.21 | 2.53 | 33.33 | 13.16 | 1.77 | 23.33 | 9.21 |
| M3 | 2.27 | 30.00 | 11.84 | 1.52 | 20.00 | 7.89 | 1.01 | 13.33 | 5.26 |
| M4 | 3.28 | 43.33 | 17.11 | 1.01 | 13.33 | 5.26 | 3.28 | 43.33 | 17.11 |
| NK1 | 3.28 | 43.33 | 17.11 | 3.28 | 43.33 | 17.11 | 1.26 | 16.67 | 6.58 |
| OX2 | 3.03 | 40.00 | 15.79 | 2.53 | 33.33 | 13.16 | 2.53 | 33.33 | 13.16 |
| P2Y1 | 1.01 | 13.33 | 5.26 | 2.78 | 36.67 | 14.47 | 2.27 | 30.00 | 11.84 |
| P2Y12 | 0.25 | 3.33 | 1.32 | 2.02 | 26.67 | 10.53 | 1.77 | 23.33 | 9.21 |
| S1P1 | 1.52 | 20.00 | 7.89 | 1.77 | 23.33 | 9.21 | 1.01 | 13.33 | 5.26 |
| SST2 | 3.54 | 46.67 | 18.42 | 2.78 | 36.67 | 14.47 | 1.01 | 13.33 | 5.26 |
| Y2 | 4.55 | 60.00 | 23.68 | 2.78 | 36.67 | 14.47 | 4.29 | 56.67 | 22.37 |
| α2A | 2.02 | 26.67 | 10.53 | 1.01 | 13.33 | 5.26 | 1.26 | 16.67 | 6.58 |
| β1 | 1.52 | 20.00 | 7.89 | 3.03 | 40.00 | 15.79 | 2.78 | 36.67 | 14.47 |
| δ | 2.53 | 33.33 | 13.16 | 2.02 | 26.67 | 10.53 | 0.25 | 3.33 | 1.32 |
| κ | 2.27 | 30.00 | 11.84 | 2.53 | 33.33 | 13.16 | 1.01 | 13.33 | 5.26 |
| μ | 1.26 | 16.67 | 6.58 | 2.53 | 33.33 | 13.16 | 2.02 | 26.67 | 10.53 |
| Mean | 2.24 | 29.58 | 11.68 | 2.42 | 31.98 | 12.62 | 1.82 | 23.96 | 9.46 |
| SD | 0.85 | 11.22 | 4.43 | 0.78 | 10.30 | 4.06 | 0.94 | 12.46 | 4.92 |
| Lower 95% CI | 1.95 | 25.69 | 10.14 | 2.15 | 28.41 | 11.22 | 1.49 | 19.64 | 7.75 |
| Upper 95% CI | 2.54 | 33.47 | 13.21 | 2.69 | 35.55 | 14.03 | 2.14 | 28.27 | 11.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhumaid, N.K.; Tawfik, E.A. Reliability of AlphaFold2 Models in Virtual Drug Screening: A Focus on Selected Class A GPCRs. Int. J. Mol. Sci. 2024, 25, 10139. https://doi.org/10.3390/ijms251810139
Alhumaid NK, Tawfik EA. Reliability of AlphaFold2 Models in Virtual Drug Screening: A Focus on Selected Class A GPCRs. International Journal of Molecular Sciences. 2024; 25(18):10139. https://doi.org/10.3390/ijms251810139
Chicago/Turabian StyleAlhumaid, Nada K., and Essam A. Tawfik. 2024. "Reliability of AlphaFold2 Models in Virtual Drug Screening: A Focus on Selected Class A GPCRs" International Journal of Molecular Sciences 25, no. 18: 10139. https://doi.org/10.3390/ijms251810139