
The Complete Mitochondrial Genome of the Siberian Scoter Melanitta stejnegeri and Its Phylogenetic Relationship in Anseriformes


Abstract
:1. Introduction
2. Results and Discussion
2.1. Mitogenome Organization
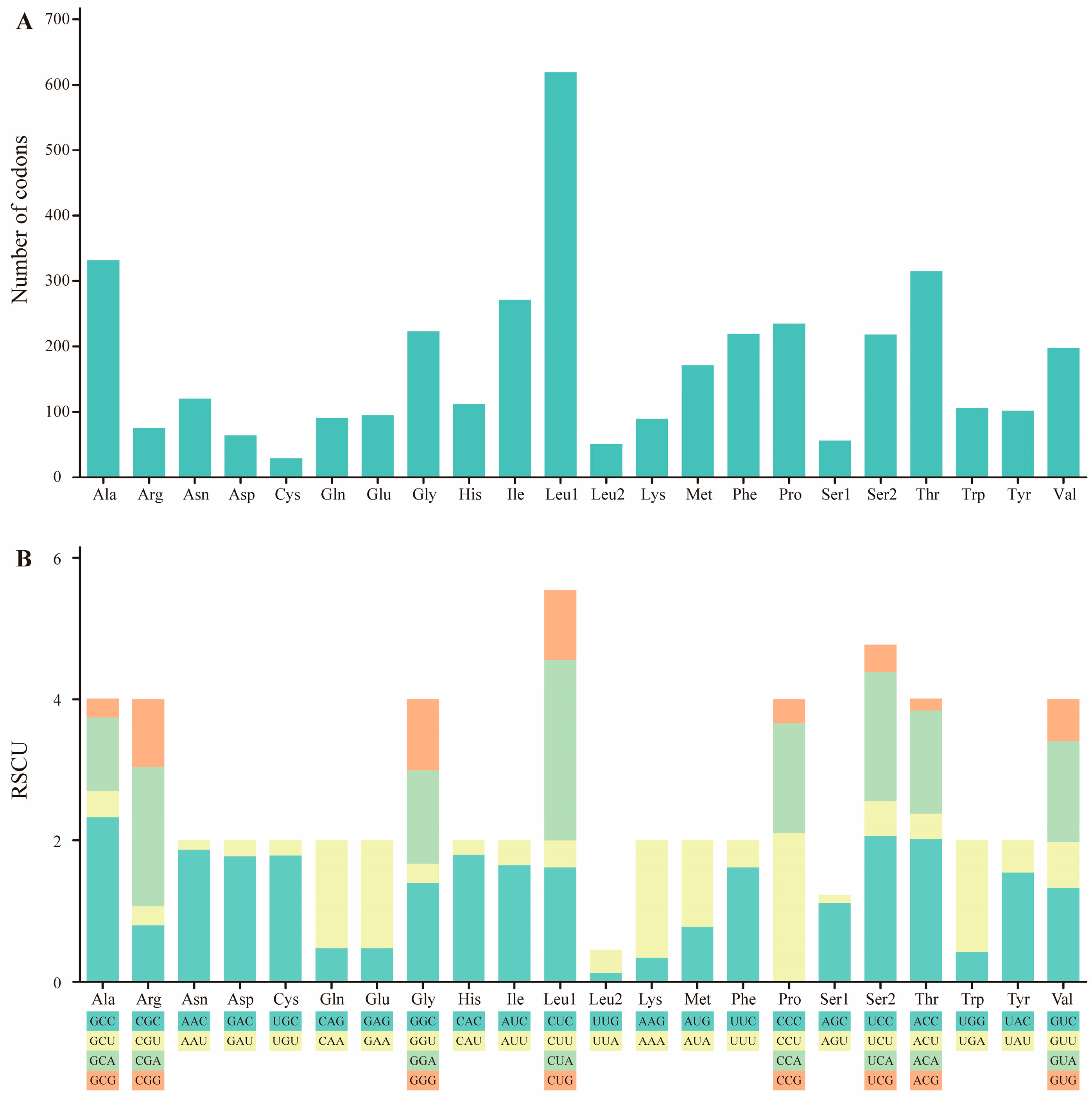
2.2. Protein-Coding Genes
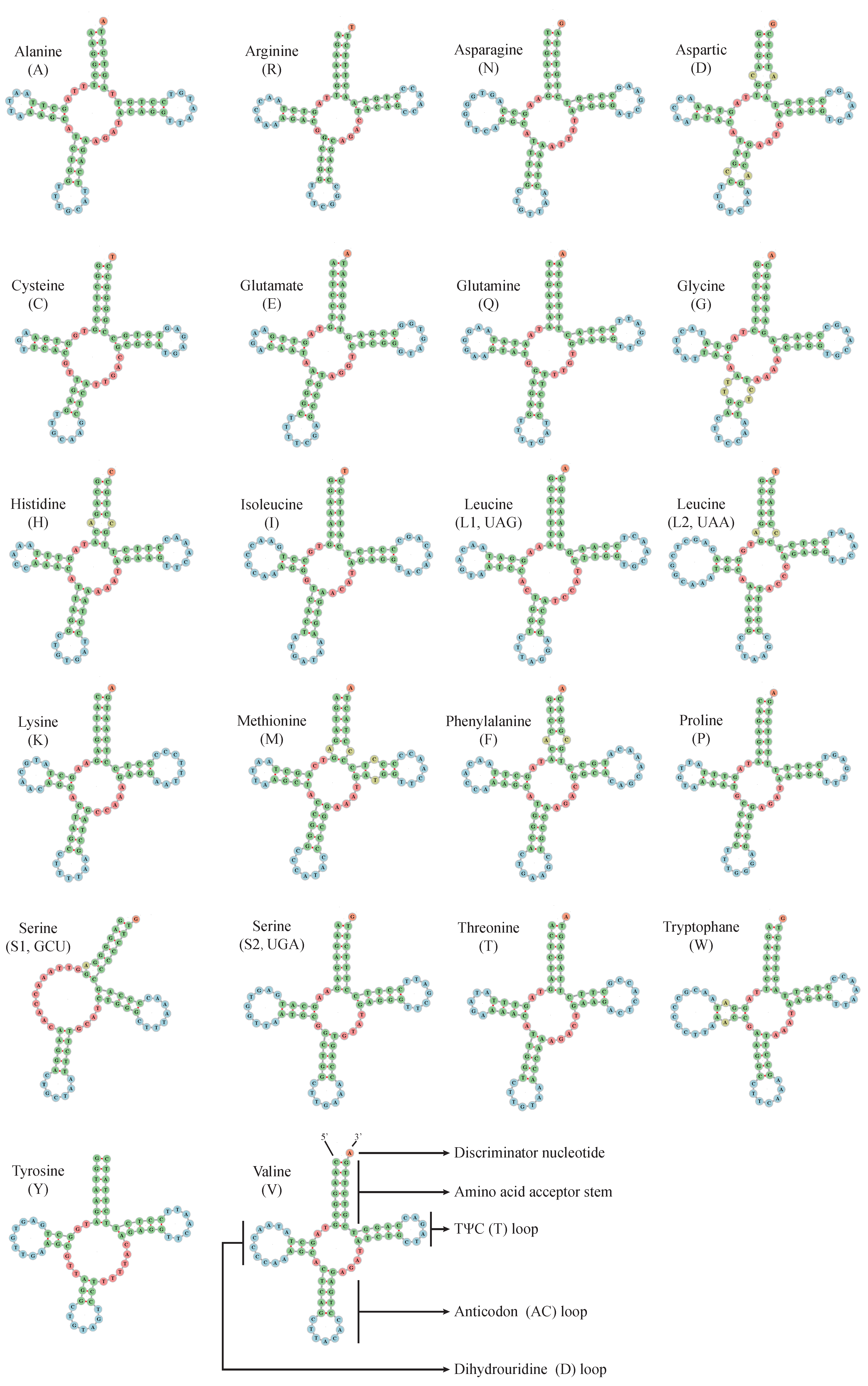
2.3. Ribosomal and Transfer RNA Genes
2.4. Non-Coding Region
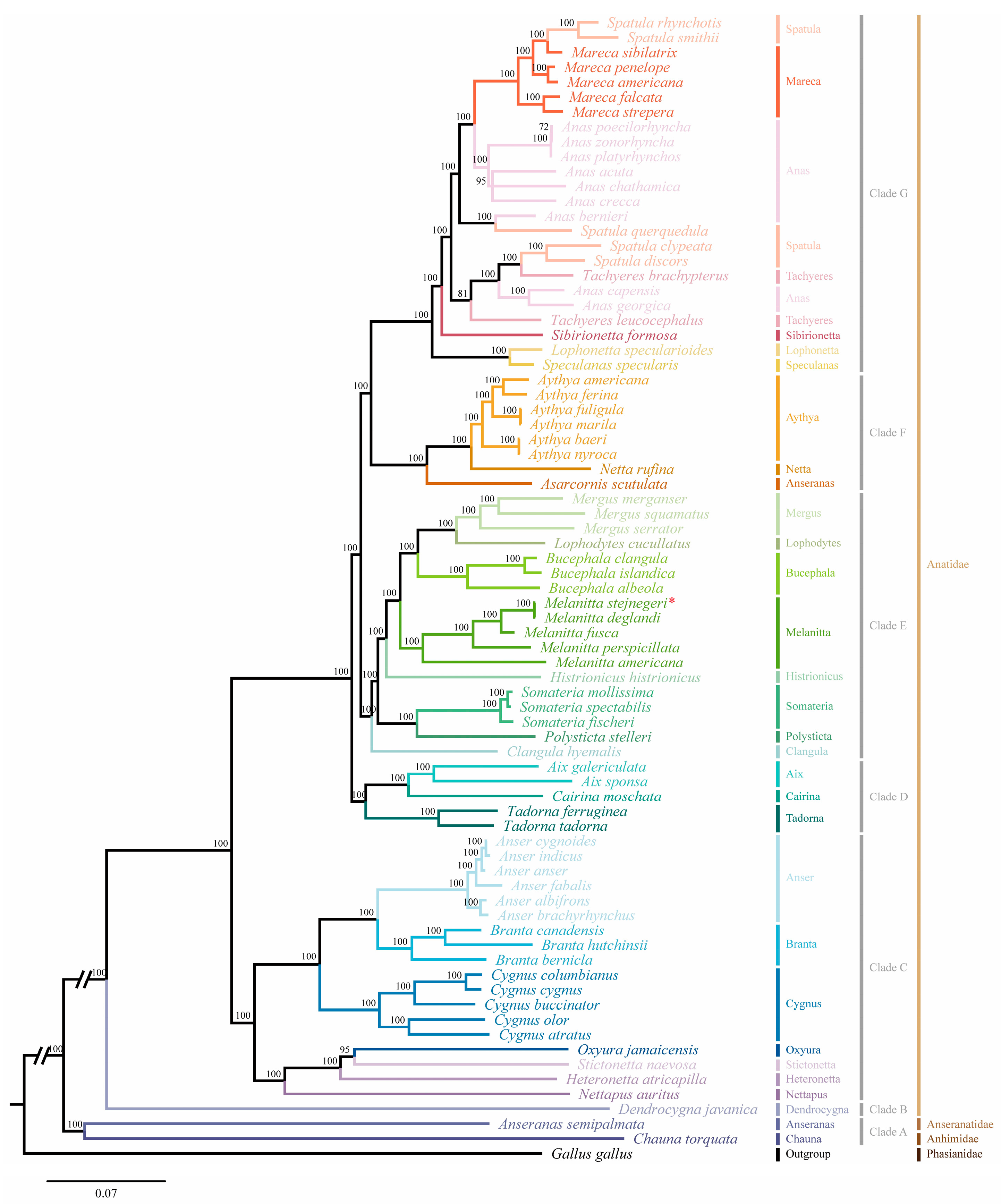
2.5. Analysis of Phylogenetic
3. Materials and Methods
3.1. Sample Collection and Mitogenome Sequencing
3.2. Assembly, Annotation, and Analysis of the Mitogenome
3.3. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, S.E.; Li, Q.; Kong, L.F.; Yu, H. Complete mitochondrial genomes of Trisidos kiyoni and Potiarca pilula: Varied mitochondrial genome size and highly rearranged gene order in Arcidae. Sci. Rep. 2016, 6, 33794. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, M.A.; Battistuzzi, F.U.; Lentino, M.; Aguilar, R.F.; Kumar, S.; Escalante, A.A. Evolution of modern birds revealed by mitogenomics Timing the radiation and origin of major orders. Mol. Biol. Evol. 2011, 28, 1927–1942. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.F.; Wang, H.; Yan, J.; Pan, T.; Jiang, S.Q.; Rao, D.Q.; Zhang, B.W. Multiple independent structural dynamic events in the evolution of snake mitochondrial genomes. BMC Genom. 2018, 19, 354. [Google Scholar] [CrossRef]
- Zhou, X.P.; Lin, Q.X.; Fang, W.Z.; Chen, X.L. The complete mitochondrial genomes of sixteen ardeid birds revealing the evolutionary process of the gene rearrangements. BMC Genom. 2014, 15, 573. [Google Scholar] [CrossRef]
- BirdLife International, Species Factsheet: Siberian Scoter Melanitta stejnegeri. Available online: https://datazone.birdlife.org/species/factsheet/siberian-scoter-melanitta-stejnegeri (accessed on 27 June 2024).
- Gill, F.; Donsker, D.; Rasmussen, P. IOC World Bird List (v14.1), 2024. Available online: https://www.worldbirdnames.org/new/ (accessed on 20 June 2024).
- Chen, P.; Li, J.Q.; Li, H.B.; Lu, Q.; Liu, W.; Zhang, J.L. Characterization of the complete mitochondrial genome of sea duck Mergus serrator and comparison with other Anseriformes species. Pak. J. Zool. 2023, 55, 2365–2376. [Google Scholar] [CrossRef]
- Sun, Z.L.; Pan, T.; Hu, C.C.; Sun, L.; Ding, H.W.; Wang, H.; Zhang, C.L.; Jin, H.; Chang, Q.; Kan, X.Z.; et al. Rapid and recent diversification patterns in Anseriformes birds: Inferred from molecular phylogeny and diversification analyses. PLoS ONE 2017, 12, e0184529. [Google Scholar] [CrossRef]
- Yuan, Z.F.; Liu, P.; Lu, X.; Zhu, D.; Liu, J.; Guo, Q.; Zhang, W.P.; Duan, Y.B. Complete mitochondrial genome and phylogenetic analysis of the blue whistling thrush (Myophonus caeruleus). Genes 2024, 15, 830. [Google Scholar] [CrossRef]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef]
- Liu, D.W.; Zhou, Y.W.; Fei, Y.L.; Xie, C.P.; Hou, S.L. Mitochondrial genome of the critically endangered Baer’s Pochard, Aythya baeri, and its phylogenetic relationship with other Anatidae species. Sci. Rep. 2021, 11, 24302. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lin, Y.F.; Xiao, Q.; Lin, Y.; Du, Y.; Lin, C.X.; Ward-Fear, G.; Hu, C.C.; Qu, Y.F.; Li, H. Characterization of the complete mitochondrial genome of the many-lined sun skink (Eutropis multifasciata) and comparison with other Scincomorpha species. Genomics 2021, 113, 2526–2536. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.L. Mitochondrial genome organization and vertebrate phylogenetics. Genet. Mol. Biol. 2000, 23, 745–752. [Google Scholar] [CrossRef]
- Steinberg, S.; Cedergren, R. Structural compensation in atypical mitochondrial tRNAs. Nat. Struct. Biol. 1994, 1, 507–510. [Google Scholar] [CrossRef]
- Kuhle, B.; Hirschi, M.; Doerfel, L.K.; Lander, G.C.; Schimmel, P. Structural basis for shape-selective recognition and aminoacylation of a D-armless human mitochondrial Trna. Nat. Commun. 2022, 13, 5100. [Google Scholar] [CrossRef]
- Biela, A.; Hammermeister, A.; Kaczmarczyk, I.; Walczak, M.; Koziej, L.; Lin, T.Y.; Glatt, S. The diverse structural modes of tRNA binding and recognition. J. Biol. Chem. 2023, 299, 104966. [Google Scholar] [CrossRef]
- Sun, C.H.; Liu, H.Y.; Min, X.; Lu, C.H. Mitogenome of the little owl Athene noctua and phylogenetic analysis of Strigidae. Int. J. Biol. Macromol. 2020, 151, 924–931. [Google Scholar] [CrossRef]
- Ruokonen, M.; Kvist, L. Structure and evolution of the avian mitochondrial control region. Mol. Phylogenet. Evol. 2002, 23, 422–432. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, Z.; Yang, C.Q.; Wang, Y.; Orr, M.C.; Wang, H.; Zhang, A.B. Identification of species by combining molecular and morphological data using convolutional neural networks. Syst. Biol. 2022, 71, 690–705. [Google Scholar] [CrossRef]
- Zou, Z.T.; Zhang, J.Z. Morphological and molecular convergences in mammalian phylogenetics. Nat. Commun. 2016, 7, 12758. [Google Scholar] [CrossRef]
- Liu, G.; Zhou, L.Z.; Li, B.; Zhang, L.L. The complete mitochondrial genome of Aix galericulata and Tadorna ferruginea Bearings on their phylogenetic position in the Anseriformes. PLoS ONE 2014, 9, e109701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.P.; Qiu, B.Y.; Zhang, D.S. Adaptive evolution analysis of mitochondrial genomes in Anseriform birds with various feeding habits. Curr. Biotechnol. 2023, 13, 748–754. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 January 2024).
- Meng, G.L.; Li, Y.Y.; Yang, C.T.; Liu, S.L. MitoZ A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic. Acids. Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3374–3376. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11 Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.Y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee In depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, 484–492. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Nylander, J. MrModeltest V2. Program distributed by the author. Bioinformatics 2004, 24, 581–583. Available online: https://www.researchgate.net/publication/285805344_MrModeltest_V2_Program_Distributed_by_the_Author (accessed on 15 January 2023).
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2 More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree v.1.4.4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 25 January 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M. stejnegeri | Size (bp) | A% | C% | T% | G% | A+T% | G+C% | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|---|
| mtDNA | 16,631 | 28.72 | 33.52 | 21.57 | 16.20 | 50.29 | 49.71 | 0.1422 | −0.3483 |
| PCGs | 11,407 | 26.25 | 34.64 | 22.84 | 16.28 | 49.08 | 50.92 | 0.0695 | −0.3605 |
| tRNA | 1546 | 29.11 | 22.32 | 26.39 | 22.19 | 55.50 | 44.50 | 0.0490 | −0.0029 |
| rRNA | 2598 | 33.26 | 27.02 | 19.36 | 20.36 | 52.62 | 47.38 | 0.2641 | −0.1405 |
| D-loop | 1071 | 26.89 | 33.05 | 23.81 | 16.25 | 50.70 | 49.30 | 0.0608 | −0.3409 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Chen, Y.; Wang, Z.; Wu, D.; Chen, P.; Chen, Y. The Complete Mitochondrial Genome of the Siberian Scoter Melanitta stejnegeri and Its Phylogenetic Relationship in Anseriformes. Int. J. Mol. Sci. 2024, 25, 10181. https://doi.org/10.3390/ijms251810181
Chen H, Chen Y, Wang Z, Wu D, Chen P, Chen Y. The Complete Mitochondrial Genome of the Siberian Scoter Melanitta stejnegeri and Its Phylogenetic Relationship in Anseriformes. International Journal of Molecular Sciences. 2024; 25(18):10181. https://doi.org/10.3390/ijms251810181
Chicago/Turabian StyleChen, Huimin, Yaqin Chen, Zhenqi Wang, Dawei Wu, Pan Chen, and Yanhong Chen. 2024. "The Complete Mitochondrial Genome of the Siberian Scoter Melanitta stejnegeri and Its Phylogenetic Relationship in Anseriformes" International Journal of Molecular Sciences 25, no. 18: 10181. https://doi.org/10.3390/ijms251810181
APA StyleChen, H., Chen, Y., Wang, Z., Wu, D., Chen, P., & Chen, Y. (2024). The Complete Mitochondrial Genome of the Siberian Scoter Melanitta stejnegeri and Its Phylogenetic Relationship in Anseriformes. International Journal of Molecular Sciences, 25(18), 10181. https://doi.org/10.3390/ijms251810181