
The Protective Role of Interleukin-37 in Cardiovascular Diseases through Ferroptosis Modulation

 ,
,  ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Ferroptosis and Cardiovascular Diseases
3. Inflammation Signaling and Ferroptosis
3.1. Damage-Associated Molecular Patterns (DAMPs) and Ferroptosis
3.2. JAK-STAT Cell Signaling Pathway and Ferroptosis
3.3. NF-κB and Ferroptosis
3.4. MAPK and Ferroptosis
3.5. cGAS-STING and Ferroptosis
3.6. NLRP3 and Ferroptosis
4. Macrophages and Ferroptosis
5. Protective Role of Interleukin-37 in Cardiovascular Diseases
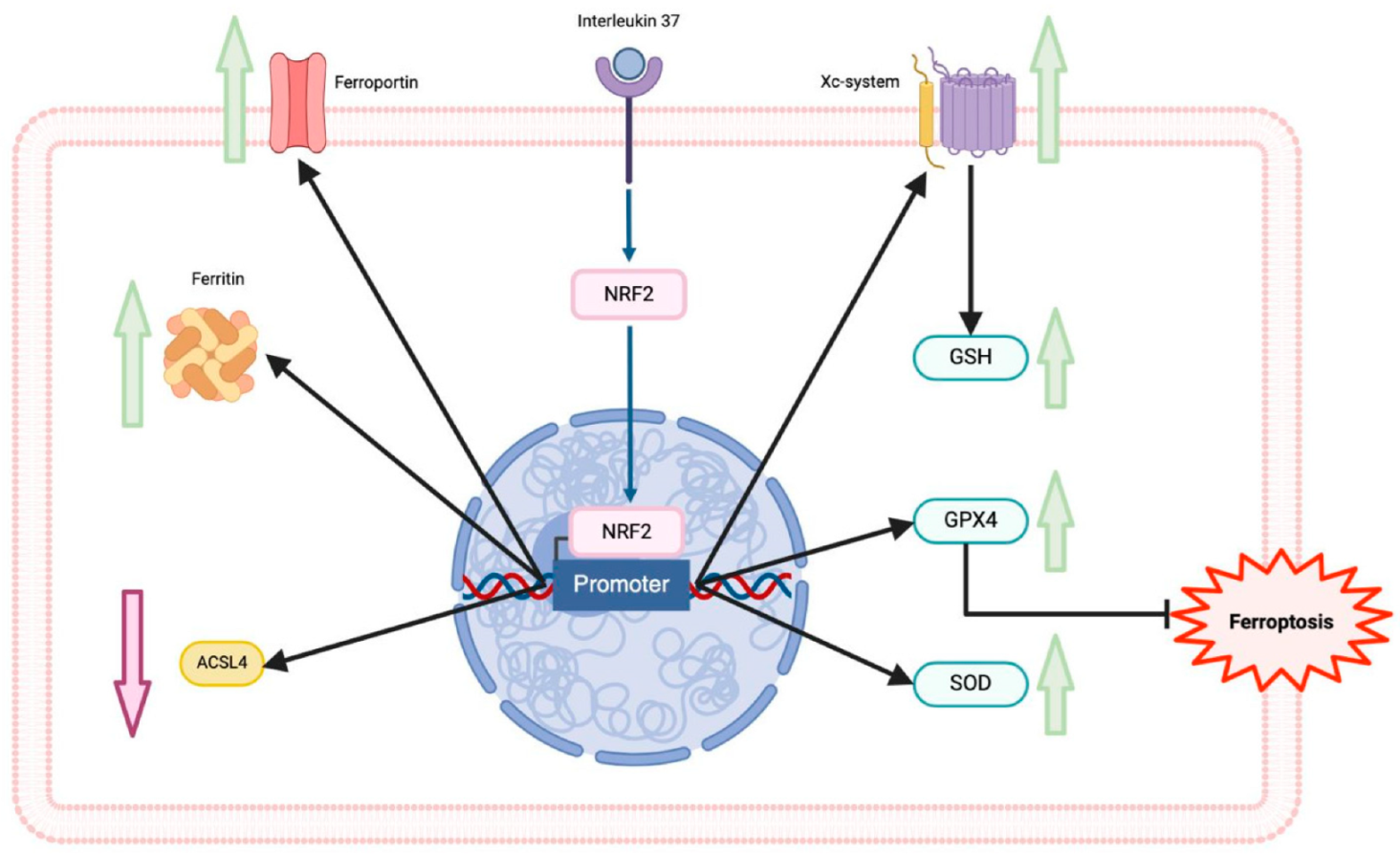
6. Interleukin-37 and Ferroptosis in Cardiovascular Diseases
7. Conclusions and Closing Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Xie, L.H.; Fefelova, N.; Pamarthi, S.H.; Gwathmey, J.K. Molecular Mechanisms of Ferroptosis and Relevance to Cardiovascular Disease. Cells 2022, 11, 2726. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Wang, S.; Miao, R.; Zhong, J. Targeting Iron Metabolism and Ferroptosis as Novel Therapeutic Approaches in Cardiovascular Diseases. Nutrients 2023, 15, 591. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, H.; Wang, S.; Tang, G.; Zhai, C.; Shen, L. Ferroptosis: A Novel Therapeutic Target for Ischemia-Reperfusion Injury. Front. Cell Dev. Biol. 2021, 9, 688605. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, A.C.; Zeng, H.; Chen, J.X. The Therapeutic Potential of Targeting Ferroptosis in the Treatment of Mitochondrial Cardiomyopathies and Heart Failure. J. Cardiovasc. Pharmacol. 2024, 83, 23–32. [Google Scholar] [CrossRef]
- Huang, T.; Wang, K.; Li, Y.; Ye, Y.; Chen, Y.; Wang, J.; Yao, C. Construction of a Novel Ferroptosis-Related Gene Signature of Atherosclerosis. Front. Cell Dev. Biol. 2022, 9, 800833. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef]
- Fonseca-Camarillo, G.; Furuzawa-Carballeda, J.; Yamamoto-Furusho, J.K. Interleukin 35 (IL-35) and IL-37: Intestinal and peripheral expression by T and B regulatory cells in patients with Inflammatory Bowel Disease. Cytokine 2015, 75, 389–402. [Google Scholar] [CrossRef]
- Law, C.C.; Puranik, R.; Fan, J.; Fei, J.; Hambly, B.D.; Bao, S. Clinical Implications of IL-32, IL-34 and IL-37 in Atherosclerosis: Speculative Role in Cardiovascular Manifestations of COVID-19. Front. Cardiovasc. Med. 2021, 8, 630767. [Google Scholar] [CrossRef]
- Xu, J.; Han, X.; Xia, N.; Zhao, Q.; Cheng, Z. IL-37 suppresses macrophage ferroptosis to attenuate diabetic atherosclerosis via the NRF2 pathway. Exp. Ther. Med. 2023, 25, 289. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Pedrera, L.; Espiritu, R.A.; Ros, U.; Weber, J.; Schmitt, A.; Stroh, J.; Hailfinger, S.; von Karstedt, S.; García-Sáez, A.J. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021, 28, 1644–1657. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- You, J.; Ouyang, S.; Xie, Z.; Zhi, C.; Yu, J.; Tan, X.; Li, P.; Lin, X.; Ma, W.; Liu, Z.; et al. The suppression of hyperlipid diet-induced ferroptosis of vascular smooth muscle cells protests against atherosclerosis independent of p53/SCL7A11/GPX4 axis. J. Cell. Physiol. 2023, 238, 1891–1908. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Y.; Lv, G.; Wang, H. Ferroptosis as a therapeutic target for inflammation-related intestinal diseases. Front. Pharmacol. 2023, 14, 1095366. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Ocansey, D.K.W.; Yuan, J.; Wei, Z.; Mao, F.; Zhang, Z. Role of ferroptosis in the pathogenesis and as a therapeutic target of inflammatory bowel disease. Int. J. Mol. Med. 2023, 51, 53. [Google Scholar] [CrossRef] [PubMed]
- Fioranelli, M.; Roccia, M.G.; Flavin, D.; Cota, L. Regulation of Inflammatory Reaction in Health and Disease. Int. J. Mol. Sci. 2021, 22, 5277. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Li, J.; Zhu, J.; Wang, R.; Xi, Q.; Wu, H.; Shi, T.; Chen, W. Astragalus polysaccharide prevents ferroptosis in a murine model of experimental colitis and human Caco-2 cells via inhibiting NRF2/HO-1 pathway. Eur. J. Pharmacol. 2021, 911, 174518. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, Z.; Xiao, J. Ferroptosis: A Double-Edged Sword in Gastrointestinal Disease. Int. J. Mol. Sci. 2021, 22, 12403. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.-W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis affects the progression of non-alcoholic steatohepatitis via the modulation of lipid peroxidation–mediated cell death in mice. Am. J. Pathol. 2019, 190, 68–81. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Agborbesong, E.; Li, L.X.; Li, L.; Li, X. Molecular Mechanisms of Epigenetic Regulation, Inflammation, and Cell Death in ADPKD. Front. Mol. Biosci. 2022, 9, 922428. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Gagliardi, M.; Cotella, D.; Santoro, C.; Corà, D.; Barlev, N.A.; Piacentini, M.; Corazzari, M. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 2019, 10, 902. [Google Scholar] [CrossRef]
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef]
- Xu, M.; Tao, J.; Yang, Y.; Tan, S.; Liu, H.; Jiang, J.; Zheng, F.; Wu, B. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis. 2020, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Colakoglu, M.; Tuncer, S.; Banerjee, S. Emerging cellular functions of the lipid metabolizing enzyme 15-Lipoxygenase-1. Cell Prolif. 2018, 51, e12472. [Google Scholar] [CrossRef]
- Uauy, R.; Mena, P.; Rojas, C. Essential fatty acid metabolism in the micropremie. Clin. Perinatol. 2000, 27, 71–93. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.I.; Benincaso, A.I.; Knoell, C.T.; Larkin, J.K.; Austen, K.F.; Robinson, D.R. Dietary omega-3 polyunsaturated fatty acids inhibit phosphoinositide formation and chemotaxis in neutrophils. J. Clin. Investig. 1993, 91, 651–660. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, H.W.; Dekkers, D.H.; Lamers, J.M. Polyunsaturated fatty acids and signalling via phospholipase C-beta and A2 in myocardium. Mol. Cell. Biochem. 1996, 157, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, J.; Song, J.; Xie, M.; Liu, Y.; Dong, Z.; Liu, X.; Li, X.; Zhang, M.; Chen, Y.; et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic. Biol. Med. 2022, 181, 130–142. [Google Scholar] [CrossRef]
- Ren, F.; Yang, Y.; Wu, K.; Zhao, T.; Shi, Y.; Song, M.; Li, J. The effects of dandelion polysaccharides on iron metabolism by regulating hepcidin via JAK/STAT signaling pathway. Oxidative Med. Cell. Longev. 2021, 2021, 7184760. [Google Scholar] [CrossRef]
- Alam, Z.; Devalaraja, S.; Li, M.; To, T.K.J.; Folkert, I.W.; Mitchell-Velasquez, E.; Dang, M.T.; Young, P.; Wilbur, C.J.; Silverman, M.A.; et al. Counter regulation of spic by NF-κB and STAT signaling controls inflammation and iron metabolism in macrophages. Cell Rep. 2020, 31, 107825. [Google Scholar] [CrossRef]
- Liu, N.; Liang, Y.; Wei, T.; Zou, L.; Huang, X.; Kong, L.; Tang, M.; Zhang, T. The role of ferroptosis mediated by NRF2/ERK-regulated ferritinophagy in CdTe QDs induced inflammation in macrophage. J. Hazard. Mater. 2022, 436, 129043. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ezquerro, J.J.; Cuenda, A. P38 signalling pathway. Int. J. Mol. Sci. 2021, 22, 1003. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Guo, Y.; Liu, C.; Yang, Y.; Fan, X.; Yang, H.; Liu, Y.; Ma, T. Function and inhibition of P38 MAP kinase signaling: Targeting multiple inflammation diseases. Biochem. Pharmacol. 2024, 220, 115973. [Google Scholar] [CrossRef]
- Gluba-Brzózka, A.; Franczyk, B.; Rysz-Górzyńska, M.; Ławiński, J.; Rysz, J. Emerging Anti-Atherosclerotic Therapies. Int. J. Mol. Sci. 2021, 22, 12109. [Google Scholar] [CrossRef]
- Liang, J.-L.; Jin, X.-K.; Zhang, S.-M.; Huang, Q.-X.; Ji, P.; Deng, X.-C.; Cheng, S.-X.; Chen, W.-H.; Zhang, X.-Z. Specific activation of cGAS-STING pathway by nanotherapeu-tics-mediated ferroptosis evoked endogenous signaling for boosting systemic tumor immunotherapy. Sci. Bull. 2023, 68, 622–636. [Google Scholar] [CrossRef]
- Li, C.; Liu, J.; Hou, W.; Kang, R.; Tang, D. STING1 promotes ferroptosis through MFN1/2-dependent mitochondrial fusion. Front. Cell Dev. Biol. 2021, 9, 698679. [Google Scholar] [CrossRef]
- Couillin, I.; Riteau, N. STING Signaling and Sterile Inflammation. Front. Immunol. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Eaton, J.W.; Balla, G.; Balla, J. Natural history of the bruise: Formation, elimination, and biological effects of oxidized hemoglobin. Oxidative Med. Cell. Longev. 2013, 2013, 9. [Google Scholar] [CrossRef]
- Soares, M.P.; Bozza, M.T. Red alert: Labile heme is an alarmin. Curr. Opin. Immunol. 2016, 38, 94–100. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, L.; Pitzer, A.L.; Li, X.; Li, P.L.; Zhang, Y. Contribution of redox-dependent activation of endothelial Nlrp3 in-flammasomes to hyperglycemia-induced endothelial dysfunction. J. Mol. Med. 2016, 94, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Erdei, J.; Tóth, A.; Balogh, E.; Nyakundi, B.B.; Bányai, E.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxidative Med. Cell. Longev. 2018, 2018, 4310816. [Google Scholar] [CrossRef]
- Schillemans, M.; Karampini, E.; Kat, M.; Bierings, R. Exocytosis of Weibel-Palade bodies: How to unpack a vascular emergency kit. J. Thromb. Haemost. 2019, 17, 6–18. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Knapp, S. Heme and hemolysis in innate immunity: Adding insult to injury. Curr. Opin. Immunol. 2018, 50, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fang, Z.M.; Yi, X.; Wei, X.; Jiang, D.S. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. 2023, 14, 205. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Feng, Z.; Meng, F.; Huo, F.; Zhu, Y.; Qin, Y.; Gui, Y.; Zhang, H.; Lin, P.; He, Q.; Li, Y.; et al. Inhibition of ferroptosis rescues M2 macrophages and alleviates arthritis by suppressing the HMGB1/TLR4/STAT3 axis in M1 macrophages. Redox Biol. 2024, 75, 103255. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, H.; Chen, Y.; Liu, X.; Tian, J.; Shen, W. The Role of Macrophage Iron Overload and Ferroptosis in Atherosclerosis. Biomolecules 2022, 12, 1702. [Google Scholar] [CrossRef]
- Marques, L.; Negre-Salvayre, A.; Costa, L.; Canonne-Hergaux, F. Iron gene expression profile in atherogenic Mox macrophages. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 1137–1146. [Google Scholar] [CrossRef]
- Cornelissen, A.; Guo, L.; Sakamoto, A.; Virmani, R.; Finn, A.V. New insights into the role of iron in inflammation and atherosclerosis. EBioMedicine 2019, 47, 598–606. [Google Scholar] [CrossRef]
- Xiao, L.; Luo, G.; Guo, X.; Jiang, C.; Zeng, H.; Zhou, F.; Li, Y.; Yu, J.; Yao, P. Macrophage iron retention aggravates atherosclerosis: Evidence for the role of autocrine formation of hepcidin in plaque macrophages. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2020, 1865, 158531. [Google Scholar] [CrossRef]
- Lai, Y.; Dong, C. Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int. Immunol. 2016, 28, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Nold-Petry, C.; Nold, M.; Fujita, M.; Li, S.; Kim, S.; Bufler, P. Suppression of innate inflammation and immunity by interleukin-37. Eur. J. Immunol. 2016, 46, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.D.; Zhao, Y.; Liu, Y. Insights into IL-37, the role in autoimmune diseases. Autoimmun. Rev. 2015, 14, 1170–1175. [Google Scholar] [CrossRef]
- Quirk, S.; Agrawal, D.K. Immunobiology of IL-37: Mechanism of action and clinical perspectives. Expert. Rev. Clin. Immunol. 2014, 10, 1703–1709. [Google Scholar] [CrossRef]
- Kumar, S.; Hanning, C.R.; Brigham-Burke, M.R.; Rieman, D.J.; Lehr, R.; Khandekar, S.; Kirkpatrick, R.B.; Scott, G.F.; Lee, J.C.; Lynch, F.J.; et al. Interleukin-1F7B (IL-1H4/IL-1 F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine 2002, 18, 61–71. [Google Scholar] [CrossRef]
- Tete, S.; Tripodi, D.; Rosati, M.; Conti, F.; Maccauro, G.; Saggini, A.; Cianchetti, E.; Caraffa, A.; Antinolfi, P.; Toniato, E.; et al. IL-37 (IL-1F7) the newest anti-inflammatory cytokine which suppresses immune responses and inflammation. Int. J. Immunopathol. Pharmacol. 2012, 25, 31–38. [Google Scholar] [CrossRef]
- Moretti, S.; Bozza, S.; Oikonomou, V.; Renga, G.; Casagrande, A.; Iannitti, R.G.; Puccetti, M.; Garlanda, C.; Kim, S.; Li, S.; et al. IL-37 inhibits inflammasome activation and disease severity in murine aspergillosis. PLoS Pathog. 2014, 10, e1004462. [Google Scholar] [CrossRef] [PubMed]
- Kuan, R.; Agrawal, D.K.; Thankam, F.G. Treg cells in atherosclerosis. Mol. Biol. Rep. 2021, 48, 4897–4910. [Google Scholar] [CrossRef]
- Lotfy, H.; Moaaz, M.; Moaaz, M. The novel role of IL-37 to enhance the anti-inflammatory response of regulatory T cells in patients with peripheral atherosclerosis. Vascular 2020, 28, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Zeng, Q.; Huang, Y.; Shi, Y.; Lin, Y.; Lu, Z.; Meng, K.; Wu, B.; Yu, K.; Chai, M.; et al. Elevated plasma IL-37, IL-18, and IL-18BP concentrations in patients with acute coronary syndrome. Mediators Inflamm. 2014, 2014, 165742. [Google Scholar] [CrossRef] [PubMed]
- Chai, M.; Ji, Q.; Zhang, H.; Zhou, Y.; Yang, Q.; Zhou, Y.; Guo, G.; Liu, W.; Han, W.; Yang, L.; et al. The Protective Effect of Interleukin-37 on Vascular Calcification and Atherosclerosis in Apolipoprotein E-Deficient Mice with Diabetes. J. Interferon Cytokine Res. 2015, 35, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Bello, R.O.; Chin, V.K.; Isnadi, M.F.A.R.; Majid, R.A.; Abdullah, M.A.; Lee, T.Y.; Zakaria, Z.A.; Hussain, M.K.; Basir, R. The Role, Involvement and Function(s) of Interleukin-35 and Interleukin-37 in Disease Pathogenesis. Int. J. Mol. Sci. 2018, 19, 1149. [Google Scholar] [CrossRef]
- Yu, K.; Min, X.; Lin, Y.; Huang, Y.; Huang, S.; Liu, L.; Peng, Y.; Meng, K.; Li, D.; Ji, Q.; et al. Increased IL-37 concentrations in patients with arterial calcification. Clin. Chim. Acta 2016, 461, 19–24. [Google Scholar] [CrossRef]
- López-Bautista, F.; Posadas-Sánchez, R.; Vázquez-Vázquez, C.; Fragoso, J.M.; Rodríguez-Pérez, J.M.; Vargas-Alarcón, G. IL-37 Gene and Cholesterol Metabolism: Association of Polymorphisms with the Presence of Hypercholesterolemia and Cardiovascular Risk Factors. The GEA Mexican Study. Biomolecules 2020, 10, 1409. [Google Scholar] [CrossRef]
- Yin, D.; Naji, D.H.; Xia, Y.; Li, S.; Bai, Y.; Jiang, G.; Zhao, Y.; Wang, X.; Huang, Y.; Chen, S.; et al. Genomic Variant in IL-37 Confers A Significant Risk of Coronary Artery Disease. Sci. Rep. 2017, 7, 42175. [Google Scholar] [CrossRef]
- Xu, D.; Wang, A.; Jiang, F.; Hu, J.; Zhang, X. Effects of interleukin-37 on cardiac function after myocardial infarction in mice. Int. J. Clin. Exp. Pathol. 2015, 8, 5247–5251. [Google Scholar]
- Wu, B.; Meng, K.; Ji, Q.; Cheng, M.; Yu, K.; Zhao, X.; Tony, H.; Liu, Y.; Zhou, Y.; Chang, C.; et al. Interleukin-37 ameliorates myocardial ischaemia/reperfusion injury in mice. Clin. Exp. Immunol. 2014, 176, 438–451. [Google Scholar] [CrossRef]
- Tian, H.; Xiong, Y.; Zhang, Y.; Leng, Y.; Tao, J.; Li, L.; Qiu, Z.; Xia, Z. Activation of NRF2/FPN1 pathway attenuates myocardial ischemia–reperfusion injury in diabetic rats by regulating iron homeostasis and ferroptosis. Cell Stress Chaperones 2021, 27, 149–164. [Google Scholar] [CrossRef]
- Ru, Y.; Luo, Y.; Liu, D.; Huang, Q.; Zhou, X.; Linghu, M.; Luo, X.; Lv, Z.; Wu, Y.; Zhang, H.; et al. Iso-rhamnetin alleviates ferroptosis-mediated colitis by activating the NRF2/HO-1 pathway and chelating iron. Int. Immunopharmacol. 2024, 135, 112318. [Google Scholar] [CrossRef]
- Deng, X.; Chu, W.; Zhang, H.; Peng, Y. Nrf2 and Ferroptosis: A New Research Direction for Ischemic Stroke. Cell. Mol. Neurobiol. 2023, 43, 3885–3896. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Gregorio, A.; Amezcua-Guerra, L.M.; Fisher-Bautista, B.; Romero-Beltrán, A.; Fonseca-Camarillo, G. The Protective Role of Interleukin-37 in Cardiovascular Diseases through Ferroptosis Modulation. Int. J. Mol. Sci. 2024, 25, 9758. https://doi.org/10.3390/ijms25189758
Cruz-Gregorio A, Amezcua-Guerra LM, Fisher-Bautista B, Romero-Beltrán A, Fonseca-Camarillo G. The Protective Role of Interleukin-37 in Cardiovascular Diseases through Ferroptosis Modulation. International Journal of Molecular Sciences. 2024; 25(18):9758. https://doi.org/10.3390/ijms25189758
Chicago/Turabian StyleCruz-Gregorio, Alfredo, Luis M. Amezcua-Guerra, Brandon Fisher-Bautista, Abraham Romero-Beltrán, and Gabriela Fonseca-Camarillo. 2024. "The Protective Role of Interleukin-37 in Cardiovascular Diseases through Ferroptosis Modulation" International Journal of Molecular Sciences 25, no. 18: 9758. https://doi.org/10.3390/ijms25189758
APA StyleCruz-Gregorio, A., Amezcua-Guerra, L. M., Fisher-Bautista, B., Romero-Beltrán, A., & Fonseca-Camarillo, G. (2024). The Protective Role of Interleukin-37 in Cardiovascular Diseases through Ferroptosis Modulation. International Journal of Molecular Sciences, 25(18), 9758. https://doi.org/10.3390/ijms25189758