
Single-Cell RNA Sequencing Reveals an Atlas of Hezuo Pig Testis Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
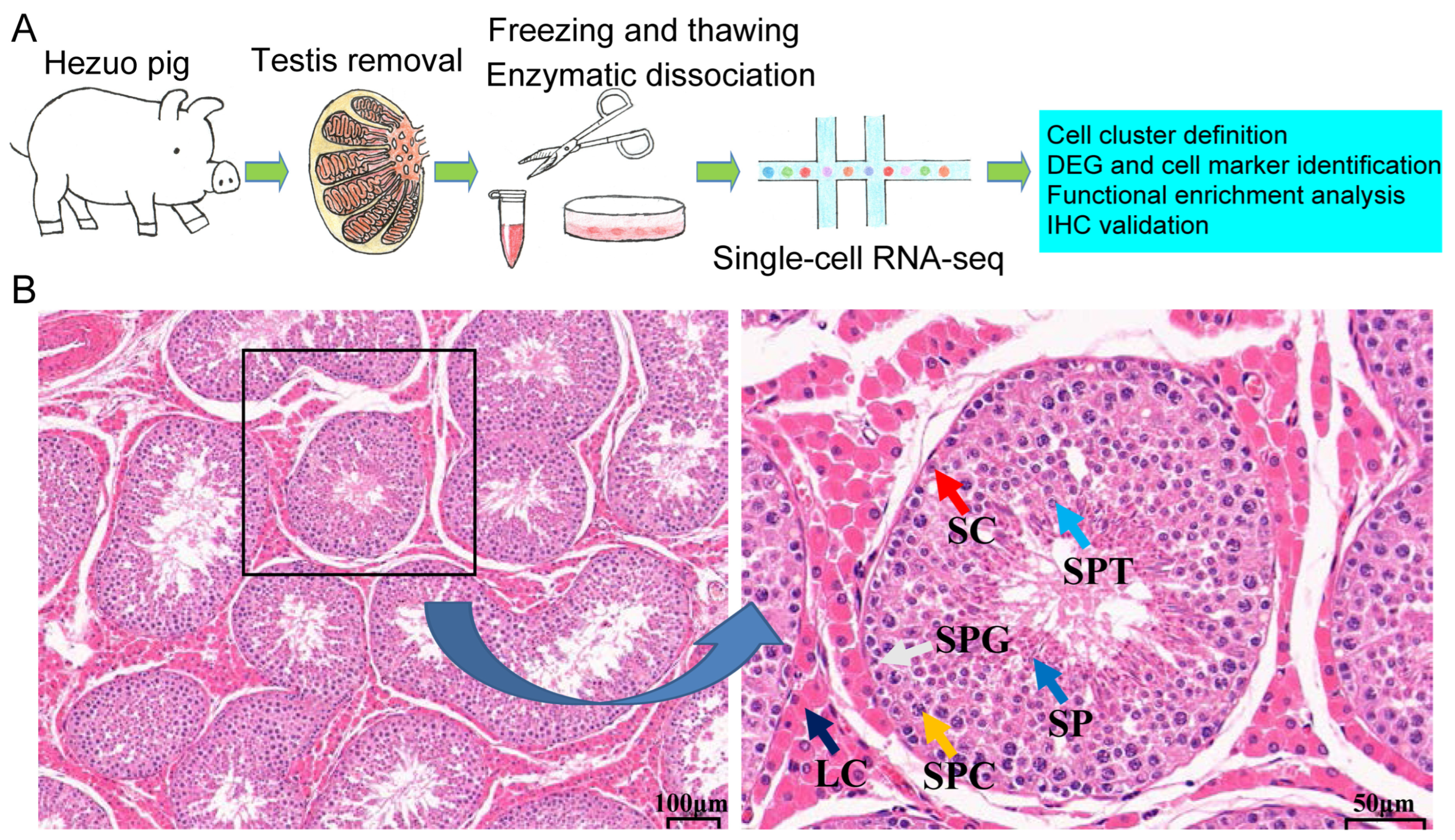
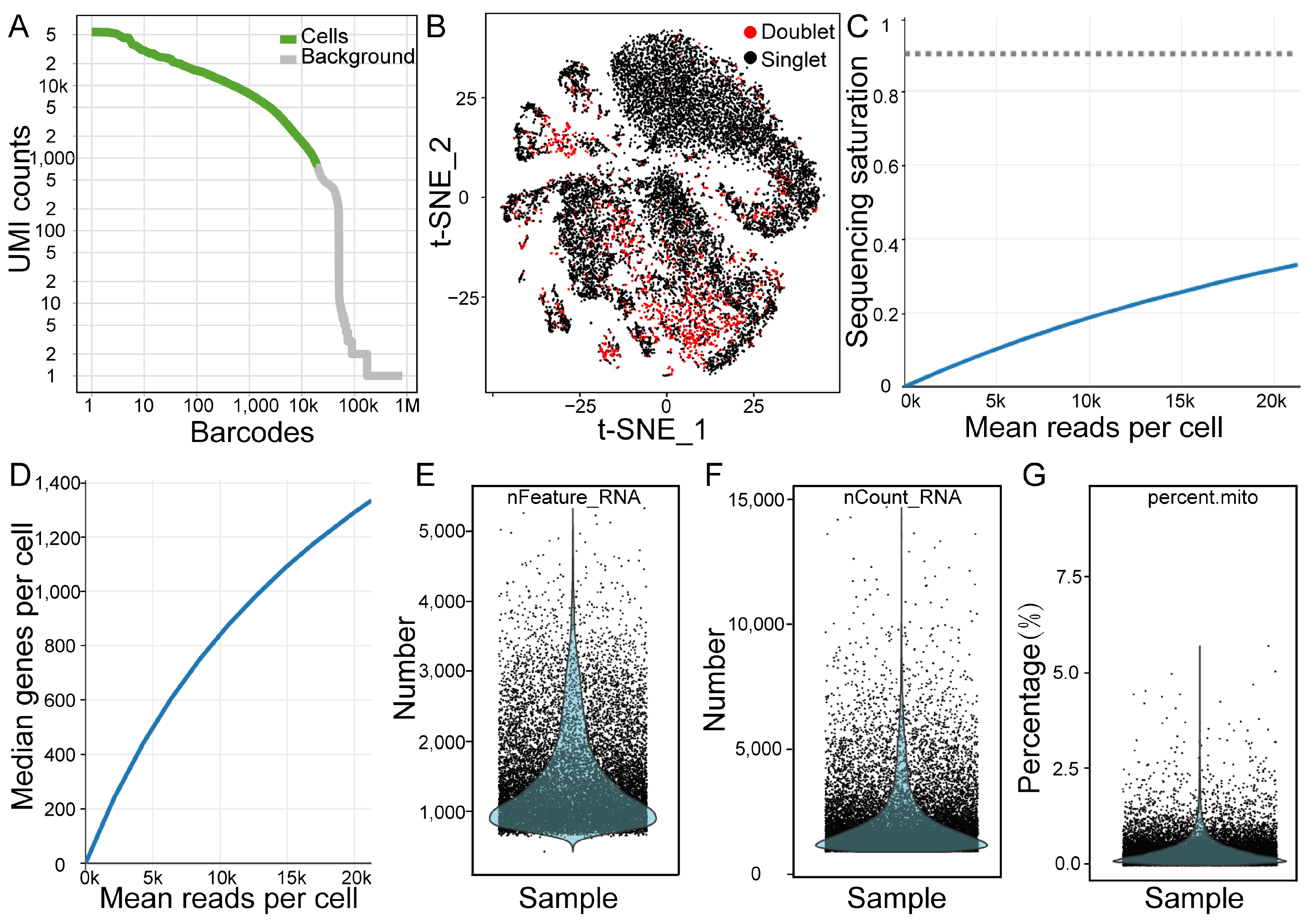
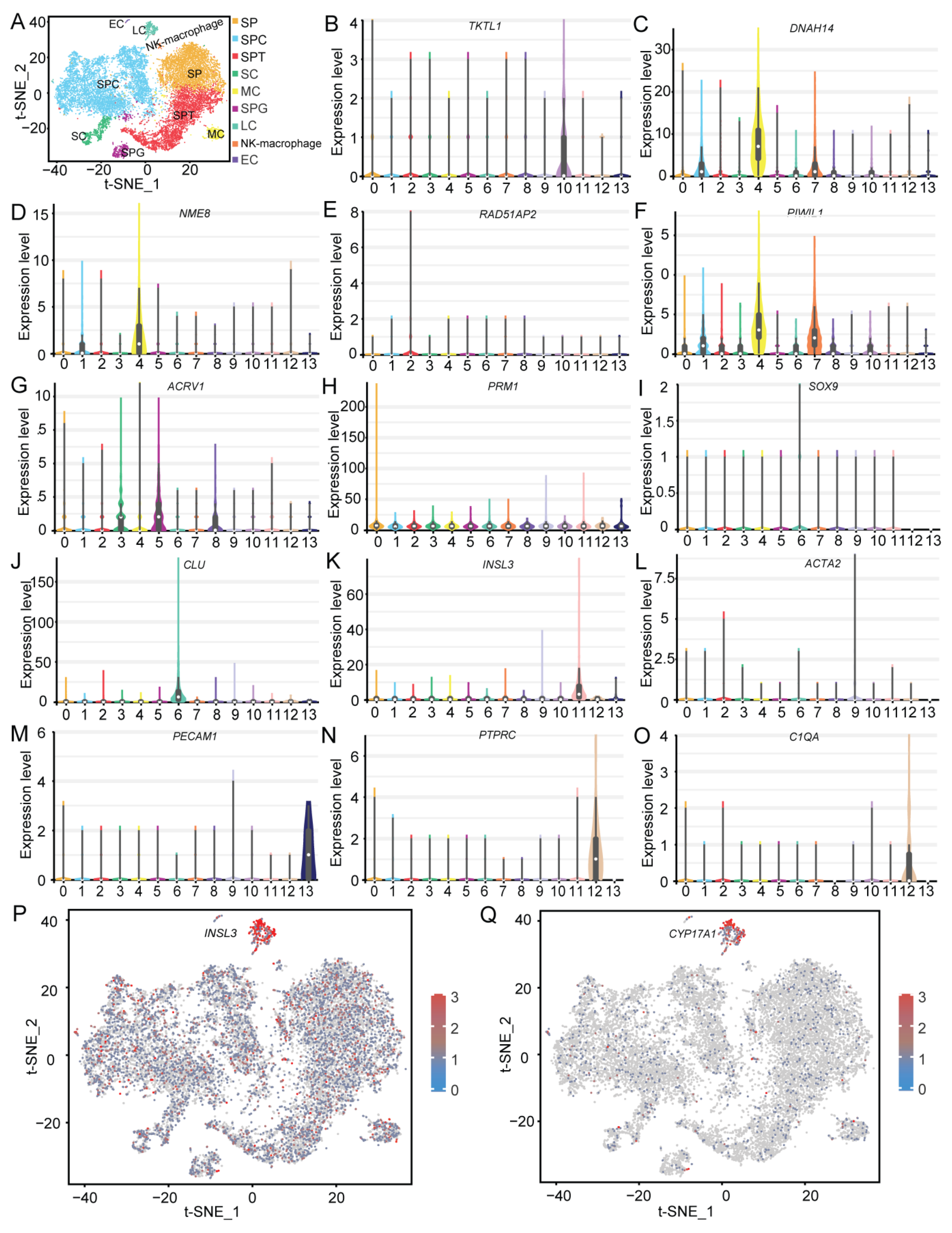
2.1. Overview of the scRNA-Seq Analysis of Testicular Cells
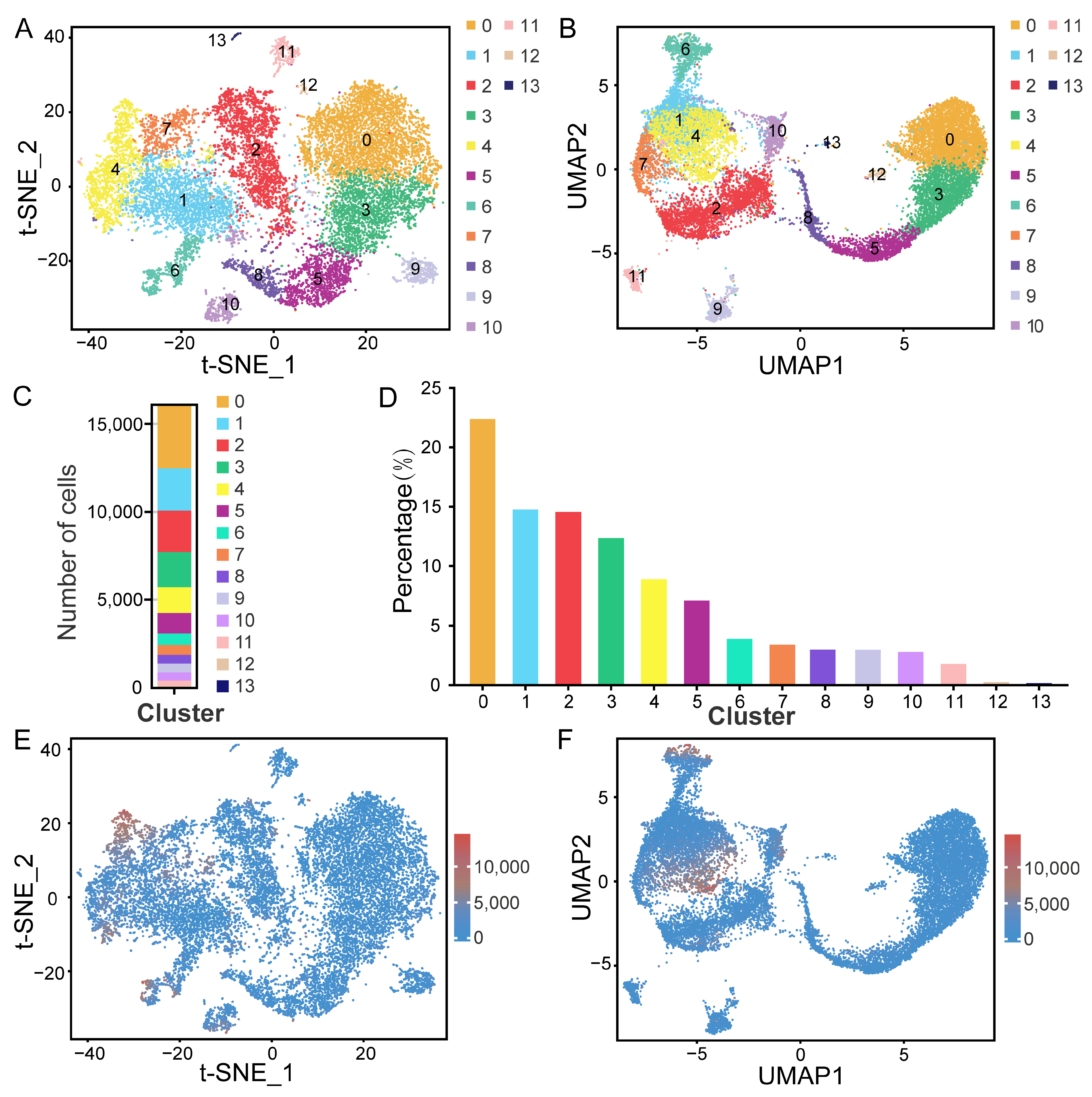
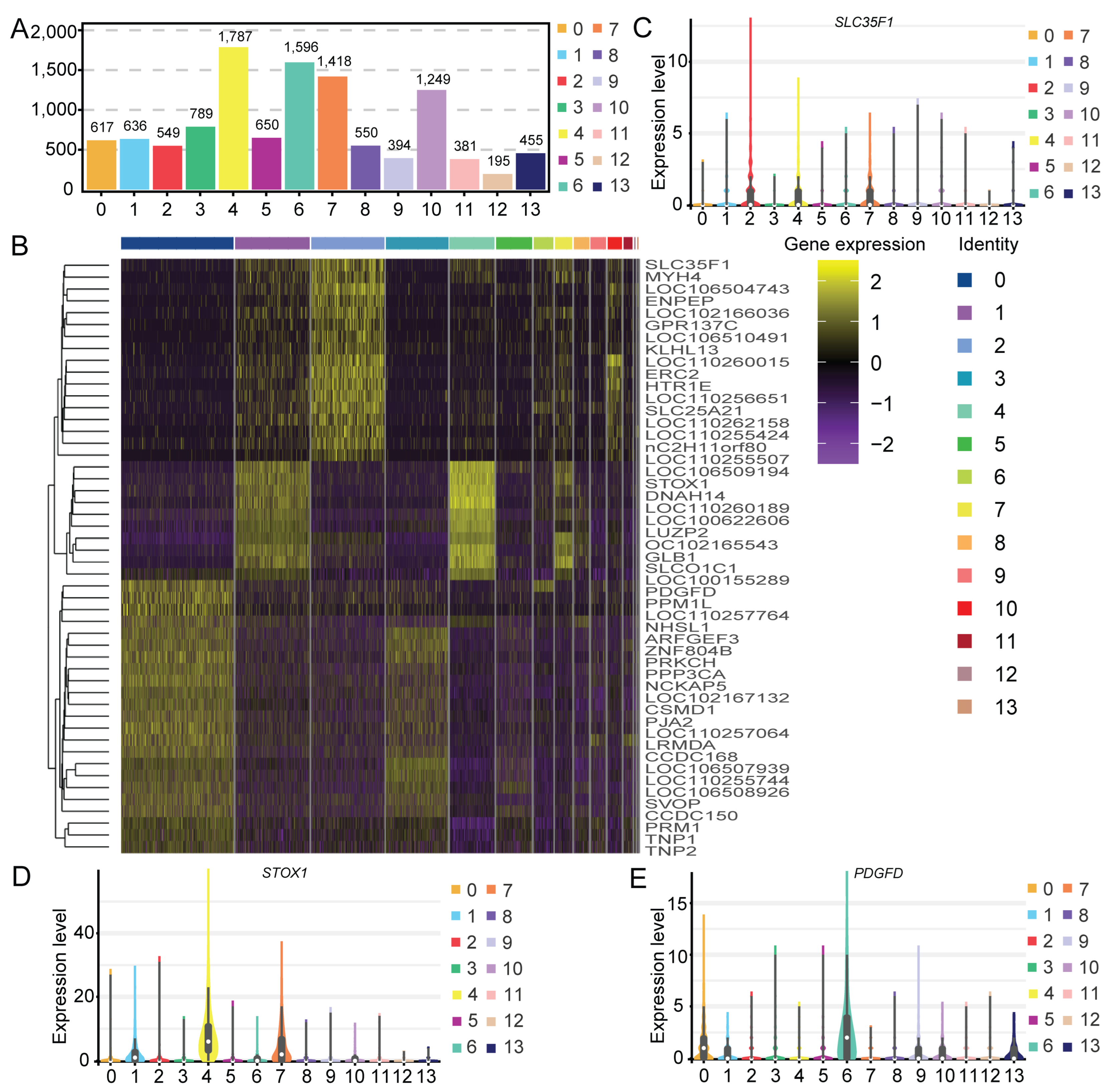
2.2. Clustering Analysis and Cell Type Identification
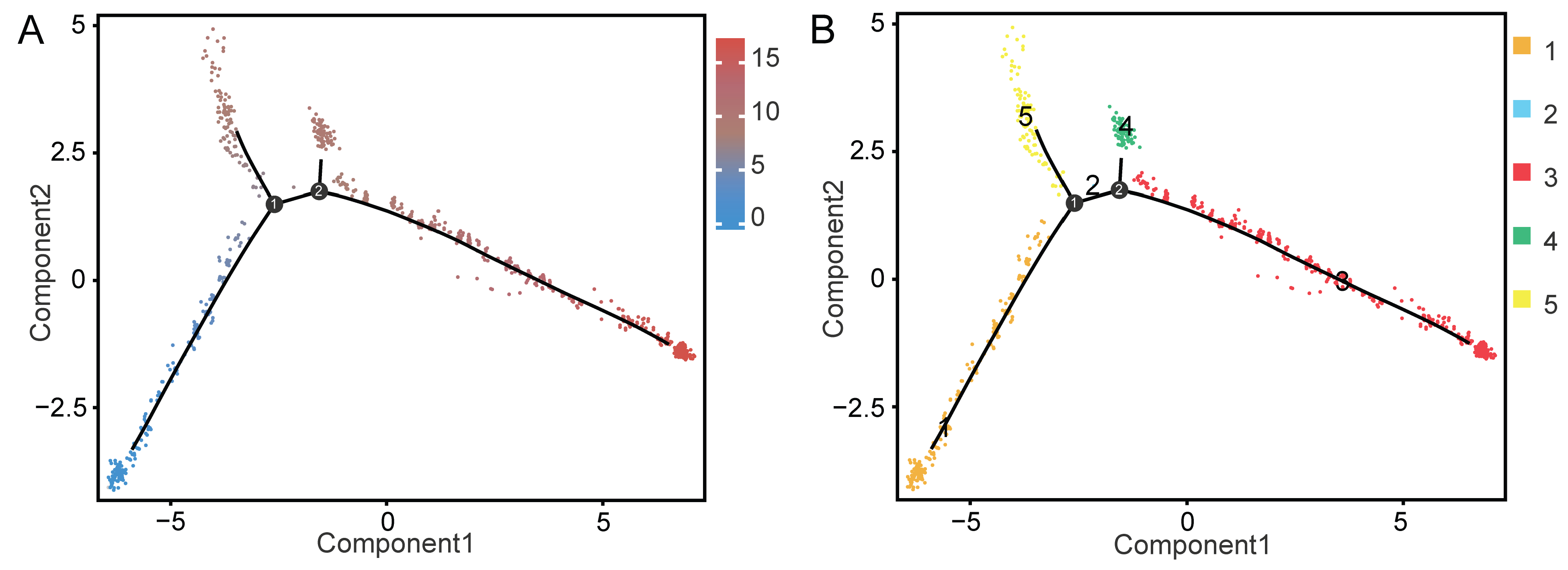
2.3. MCs and LCs Developed and Differentiated from the Same Progenitor Cells
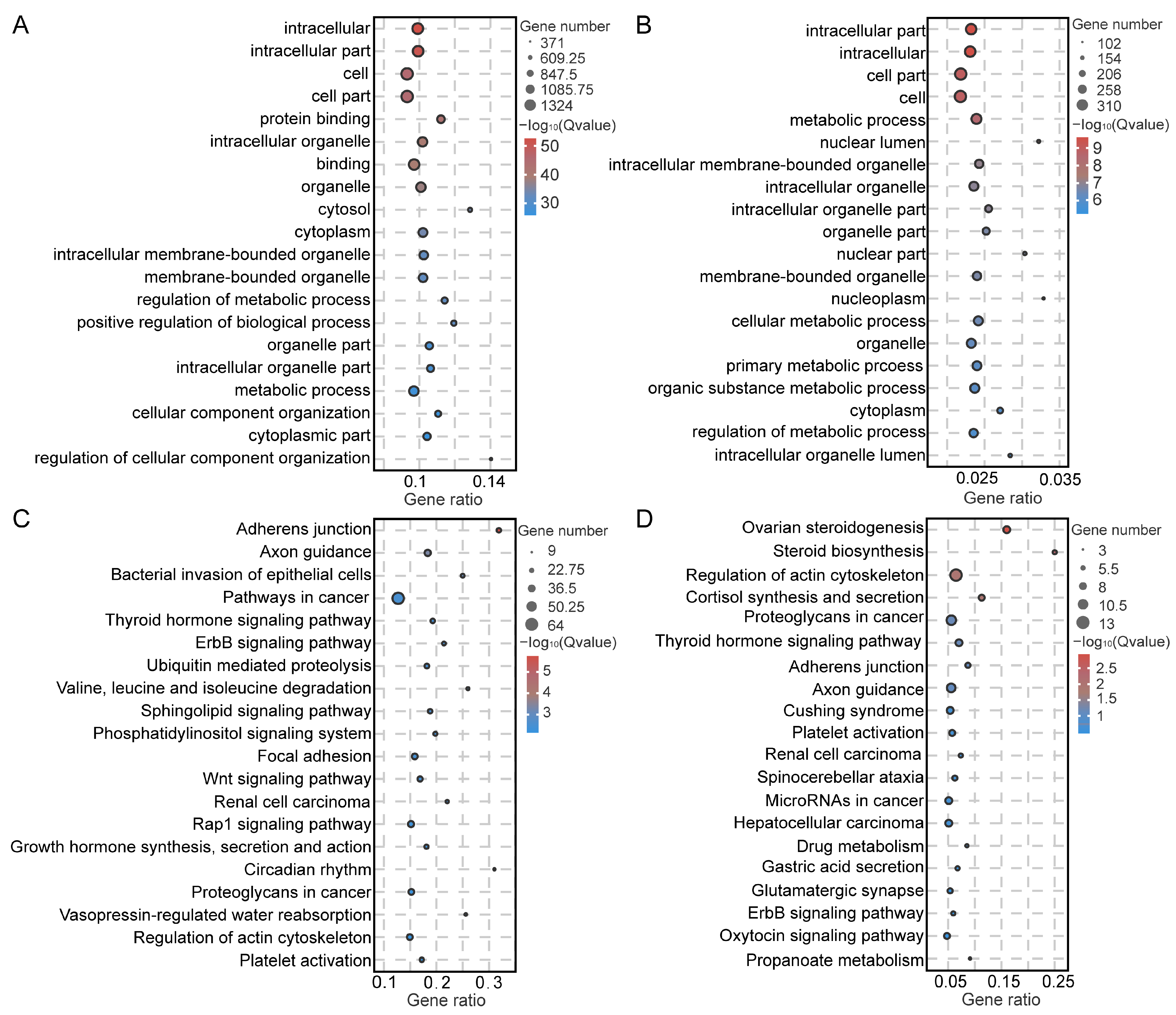
2.4. Functional Enrichment Analysis of Testicular Somatic Cells
2.5. Functional Enrichment Analysis of Testicular Germ Cells
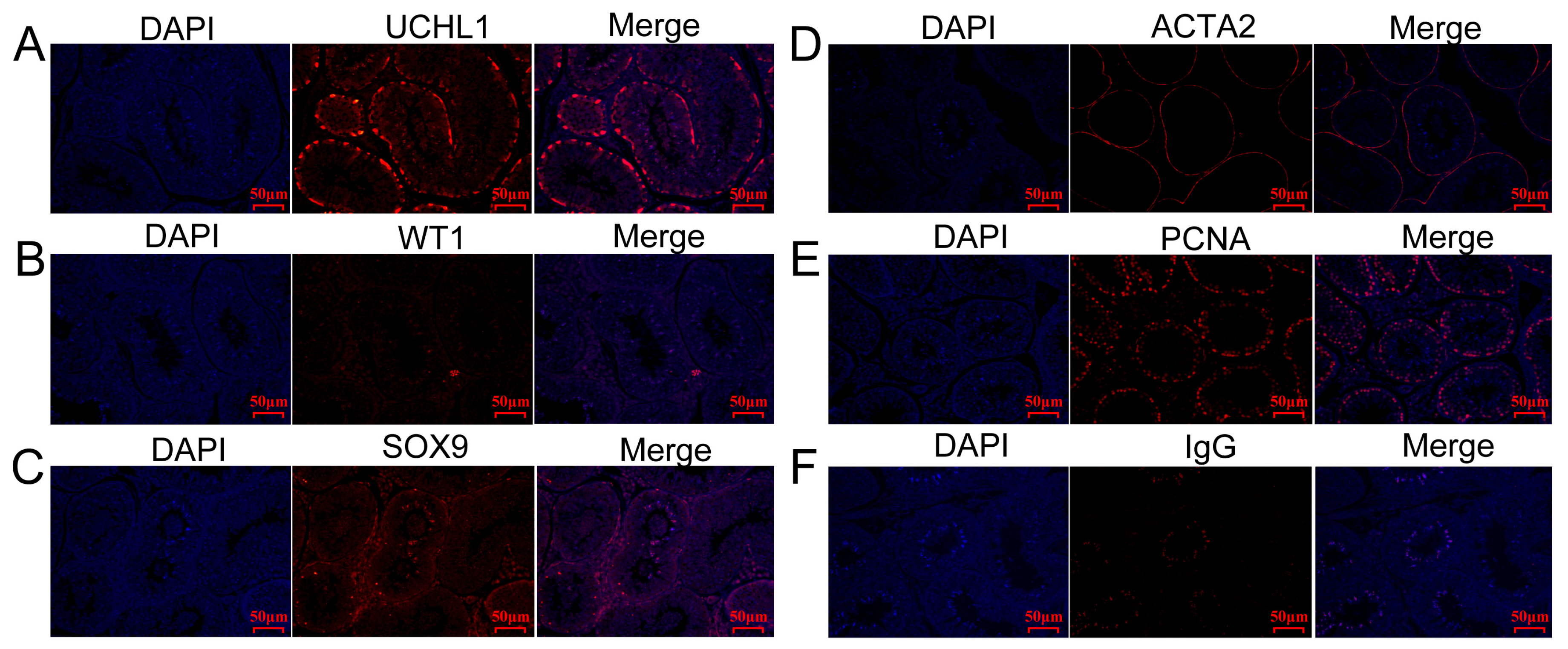
2.6. Characterization of Testicular Cells
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Testicular Sample Collection
4.3. Testicular Single-Cell Suspension Acquisition
4.4. 10× Genomics Library Preparation and Sequencing
4.5. Genome Alignment and Gene Expression Quantification
4.6. Cell Clustering
4.7. DEG Analysis and Annotation
4.8. Cell Trajectory Analysis
4.9. Hematoxylin and Eosin (H&E) Staining and Immunohistochemical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, J.; Wang, P.; Yan, Z.; Yang, Q.; Huang, X.; Gao, X.; Li, J.; Wang, Z.; Gao, Y.; Gun, S. Cloning of STC-1 and analysis of its differential expression in Hezuo pig. Anim. Biotechnol. 2023, 34, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhang, B.; Shi, H.; Yan, Z.; Wang, P.; Yang, Q.; Huang, X.; Li, J.; Wang, Z.; Gun, S. Cloning, expression analysis and localization of DAZL gene implicated in germ cell development of male Hezuo pig. Anim. Biotechnol. 2023, 34, 4000–4014. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Song, K.; Wang, P.; Gun, S.; Long, X. Evaluation of the Genetic Diversity and Population Structure of Four Native Pig Populations in Gansu Province. Int. J. Mol. Sci. 2023, 24, 17154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yan, Z.; Wang, P.; Yang, Q.; Huang, X.; Shi, H.; Tang, Y.; Ji, Y.; Zhang, J.; Gun, S. Identification and Characterization of lncRNA and mRNA in Testes of Landrace and Hezuo Boars. Animals 2021, 11, 2263. [Google Scholar] [CrossRef]
- Du, H.; Yan, Z.; Shi, H.; Yang, Q.; Huang, X.; Wang, P.; Gao, X.; Yang, J.; Gun, S. Isolation, culture, characterization and the functional study of testicular Leydig cells from Hezuo pig. Reprod. Domest. Anim. 2024, 59, e14583. [Google Scholar] [CrossRef]
- Yin, X.; Wang, P.; Yan, Z.; Yang, Q.; Huang, X.; Gun, S. Effects of Whole-Plant Corn Silage on Growth Performance, Serum Biochemical Indices, and Fecal Microorganisms in Hezuo Pigs. Animals 2024, 14, 662. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, B.; Shi, H.; Yan, Z.; Wang, P.; Yang, Q.; Huang, X.; Gun, S. Molecular characterization, expression patterns and cellular localization of BCAS2 gene in male Hezuo pig. PeerJ 2023, 11, e16341. [Google Scholar] [CrossRef]
- Wang, L.; Wang, P.; Yan, Z.; Zhang, P.; Yin, X.; Jia, R.; Li, Y.; Yang, J.; Gun, S.; Yang, Q. Whole-plant silage maize to improve fiber digestive characteristics and intestinal microbiota of Hezuo pigs. Front. Microbiol. 2024, 15, 1360505. [Google Scholar] [CrossRef]
- Zhang, B.; Yan, Z.; Gao, Y.; Li, J.; Wang, Z.; Wang, P.; Yang, Q.; Huang, X.; Gun, S. Integrated analysis of miRNA and mRNA expression profiles in testes of Landrace and Hezuo boars. Front. Vet. Sci. 2022, 9, 942669. [Google Scholar] [CrossRef]
- Li, L.; Lin, W.; Wang, Z.; Huang, R.; Xia, H.; Li, Z.; Deng, J.; Ye, T.; Huang, Y.; Yang, Y. Hormone Regulation in Testicular Development and Function. Int. J. Mol. Sci. 2024, 25, 5805. [Google Scholar] [CrossRef]
- Karimi, H.; Saraskanroud, M.R.; Koucheh, F.B. Influence of laterality on testis anatomy and histology in Ghezel rams. Vet. Med. Sci. 2019, 5, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.J.; DeFalco, T. Role of the testis interstitial compartment in spermatogonial stem cell function. Reproduction 2017, 153, R151–R162. [Google Scholar] [CrossRef] [PubMed]
- Fayomi, A.P.; Orwig, K.E. Spermatogonial stem cells and spermatogenesis in mice, monkeys and men. Stem Cell Res. 2018, 29, 207–214. [Google Scholar] [CrossRef]
- Törzsök, P.; Steiner, C.; Pallauf, M.; Abenhardt, M.; Milinovic, L.; Plank, B.; Rückl, A.; Sieberer, M.; Lusuardi, L.; Deininger, S. Long-Term Follow-Up after Testicular Torsion: Prospective Evaluation of Endocrine and Exocrine Testicular Function, Fertility, Oxidative Stress and Erectile Function. J. Clin. Med. 2022, 11, 6507. [Google Scholar] [CrossRef]
- Takashima, S. Biology and manipulation technologies of male germline stem cells in mammals. Reprod. Med. Biol. 2018, 17, 398–406. [Google Scholar] [CrossRef]
- Caires, K.; Broady, J.; McLean, D. Maintaining the male germline: Regulation of spermatogonial stem cells. J. Endocrinol. 2010, 205, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, J.-A.; Koskenniemi, J.J.; Virtanen, H.E.; Toppari, J. Testis Development. Endocr. Rev. 2018, 40, 857–905. [Google Scholar] [CrossRef]
- Ramm, S.A.; Schärer, L.; Ehmcke, J.; Wistuba, J. Sperm competition and the evolution of spermatogenesis. Mol. Hum. Reprod. 2014, 20, 1169–1179. [Google Scholar] [CrossRef]
- Wang, X.; Pei, J.; Xiong, L.; Guo, S.; Cao, M.; Kang, Y.; Ding, Z.; La, Y.; Liang, C.; Yan, P.; et al. Single-Cell RNA Sequencing Reveals Atlas of Yak Testis Cells. Int. J. Mol. Sci. 2023, 24, 7982. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Y. Action and Interaction between Retinoic Acid Signaling and Blood–Testis Barrier Function in the Spermatogenesis Cycle. Cells 2022, 11, 352. [Google Scholar] [CrossRef]
- Wanjari, U.R.; Gopalakrishnan, A.V. Blood-testis barrier: A review on regulators in maintaining cell junction integrity between Sertoli cells. Cell Tissue Res. 2024, 396, 157–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ma, J.; Wan, Z.; Wang, Q.; Wang, Z.; Zhao, J.; Wang, F.; Zhang, Y. Characterization of sheep spermatogenesis through single-cell RNA sequencing. FASEB J. 2020, 35, e21187. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T. Overview of single-cell RNA sequencing analysis and its application to spermatogenesis research. Reprod. Med. Biol. 2023, 22, e12502. [Google Scholar] [CrossRef] [PubMed]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2017, 18, 35–45. [Google Scholar] [CrossRef]
- Duhan, L.; Kumari, D.; Naime, M.; Parmar, V.S.; Chhillar, A.K.; Dangi, M.; Pasrija, R. Single-cell transcriptomics: Background, technologies, applications, and challenges. Mol. Biol. Rep. 2024, 51, 600. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The Technology and Biology of Single-Cell RNA Sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, Y.; Guan, M.; Liu, M.; Sun, S. Single-cell and spatial transcriptomic investigation reveals the spatiotemporal specificity of the beta-defensin gene family during mouse sperm maturation. Cell Commun. Signal. 2024, 22, 267. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, J.; Zhang, P.; Huang, X.; Yang, W.; Liu, R.; Sun, Q.; Lu, Y.; Zhang, M.; Fu, Q. Single-cell RNA sequencing uncovers dynamic roadmap and cell-cell communication during buffalo spermatogenesis. iScience 2022, 26, 105733. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pei, J.; Xiong, L.; Kang, Y.; Guo, S.; Cao, M.; Ding, Z.; Bao, P.; Chu, M.; Liang, C.; et al. Single-cell RNA sequencing and UPHLC-MS/MS targeted metabolomics offer new insights into the etiological basis for male cattle-yak sterility. Int. J. Biol. Macromol. 2023, 253, 126831. [Google Scholar] [CrossRef]
- Yu, X.-W.; Li, T.-T.; Du, X.-M.; Shen, Q.-Y.; Zhang, M.-F.; Wei, Y.-D.; Yang, D.-H.; Xu, W.-J.; Chen, W.-B.; Bai, C.-L.; et al. Single-cell RNA sequencing reveals atlas of dairy goat testis cells. Zool. Res. 2021, 42, 401–405. [Google Scholar] [CrossRef]
- Ren, F.; Xi, H.; Qiao, P.; Li, Y.; Xian, M.; Zhu, D.; Hu, J. Single-cell transcriptomics reveals male germ cells and Sertoli cells developmental patterns in dairy goats. Front. Cell Dev. Biol. 2022, 10, 944325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, M.; Liu, Z.; Liu, R.; Zheng, Y.; Yu, T.; Lv, Y.; Lu, H.; Zeng, W.; Zhang, T.; et al. Single-cell RNA-seq analysis of testicular somatic cell development in pigs. J. Genet. Genom. 2022, 49, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, F.; Lei, P.; Guo, M.; Liu, R.; Wang, L.; Yu, T.; Lv, Y.; Zhang, T.; Zeng, W.; et al. Single-cell RNA-sequencing reveals the dynamic process and novel markers in porcine spermatogenesis. J. Anim. Sci. Biotechnol. 2021, 12, 122. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, T.; Li, J.; Niu, C.; Sun, W.; Zhu, S.; Zhao, H.; Qiao, G.; Han, M.; He, X.; et al. The Transcriptional Cell Atlas of Testis Development in Sheep at Pre-Sexual Maturity. Curr. Issues Mol. Biol. 2022, 44, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, Y.; Hou, R.; Shi, K.; Chen, Y.; Feng, T.; An, J. Single-cell RNA-sequencing analysis and characterisation of testicular cells in giant panda (Ailuropoda melanoleuca). Reprod. Fertil. Dev. 2022, 34, 933–943. [Google Scholar] [CrossRef]
- Sohni, A.; Tan, K.; Song, H.-W.; Burow, D.; de Rooij, D.G.; Laurent, L.; Hsieh, T.-C.; Rabah, R.; Hammoud, S.S.; Vicini, E.; et al. The Neonatal and Adult Human Testis Defined at the Single-Cell Level. Cell Rep. 2019, 26, 1501–1517.e4. [Google Scholar] [CrossRef]
- Guo, J.; Nie, X.; Giebler, M.; Mlcochova, H.; Wang, Y.; Grow, E.J.; DonorConnect; Kim, R.; Tharmalingam, M.; Matilionyte, G.; et al. The dynamic transcriptional cell atlas of testis development during human puberty. Cell Stem Cell 2020, 26, 262–276. [Google Scholar] [CrossRef]
- White-Cooper, H.; Bausek, N. Evolution and spermatogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1465–1480. [Google Scholar] [CrossRef]
- Qu, N.; Ogawa, Y.; Kuramasu, M.; Nagahori, K.; Sakabe, K.; Itoh, M. Immunological microenvironment in the testis. Reprod. Med. Biol. 2019, 19, 24–31. [Google Scholar] [CrossRef]
- Figueiredo, M.G.; Gagliano-Jucá, T.; Basaria, S. Male Reproduction and Aging. Endocrinol. Metab. Clin. N. Am. 2023, 52, 211–228. [Google Scholar] [CrossRef]
- Kulibin, A.Y.; Malolina, E.A. The Rete Testis: Development and Role in Testis Function. Russ. J. Dev. Biol. 2021, 52, 370–378. [Google Scholar] [CrossRef]
- O’donnell, L.; Smith, L.B.; Rebourcet, D. Sertoli cells as key drivers of testis function. Semin. Cell Dev. Biol. 2022, 121, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Navin, N.E. Advances and Applications of Single-Cell Sequencing Technologies. Mol. Cell 2015, 58, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Kim, J.K. Dissecting Cellular Heterogeneity Using Single-Cell RNA Sequencing. Mol. Cells 2019, 42, 189–199. [Google Scholar] [CrossRef]
- Dong, F.; Ping, P.; Ma, Y.; Chen, X.-F. Application of single-cell RNA sequencing on human testicular samples: A comprehensive review. Int. J. Biol. Sci. 2023, 19, 2167–2197. [Google Scholar] [CrossRef]
- Xue, R.; Li, R.; Bai, F. Single cell sequencing: Technique, application, and future development. Sci. Bull. 2015, 60, 33–42. [Google Scholar] [CrossRef]
- Tan, K.; Wilkinson, M.F. A single-cell view of spermatogonial stem cells. Curr. Opin. Cell Biol. 2020, 67, 71–78. [Google Scholar] [CrossRef]
- Walker, W.H. Molecular mechanisms of testosterone action in spermatogenesis. Steroids 2009, 74, 602–607. [Google Scholar] [CrossRef]
- Toocheck, C.; Clister, T.; Shupe, J.; Crum, C.; Ravindranathan, P.; Lee, T.-K.; Ahn, J.-M.; Raj, G.V.; Sukhwani, M.; Orwig, K.E.; et al. Mouse Spermatogenesis Requires Classical and Nonclassical Testosterone Signaling. Biol. Reprod. 2016, 94, 11. [Google Scholar] [CrossRef]
- Zhou, R.; Wu, J.; Liu, B.; Jiang, Y.; Chen, W.; Li, J.; He, Q.; He, Z. The roles and mechanisms of Leydig cells and myoid cells in regulating spermatogenesis. Cell. Mol. Life Sci. 2019, 76, 2681–2695. [Google Scholar] [CrossRef]
- Morrison, S.J.; Spradling, A.C. Stem Cells and Niches: Mechanisms That Promote Stem Cell Maintenance throughout Life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Hou, J.; Liu, Y.; Liu, Y.; Yang, H.; Li, Z.; He, Z. The roles and regulation of Sertoli cells in fate determinations of spermatogonial stem cells and spermatogenesis. Semin. Cell Dev. Biol. 2014, 29, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Griswold, M.D. The central role of Sertoli cells in spermatogenesis. Semin. Cell Dev. Biol. 1998, 9, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Rebourcet, D.; O’shaughnessy, P.J.; Monteiro, A.; Milne, L.; Cruickshanks, L.; Jeffrey, N.; Guillou, F.; Freeman, T.C.; Mitchell, R.T.; Smith, L.B. Sertoli Cells Maintain Leydig Cell Number and Peritubular Myoid Cell Activity in the Adult Mouse Testis. PLoS ONE 2014, 9, e105687. [Google Scholar] [CrossRef]
- Chen, Q.; Deng, T.; Han, D. Testicular immunoregulation and spermatogenesis. Semin. Cell Dev. Biol. 2016, 59, 157–165. [Google Scholar] [CrossRef]
- Lau, X.; Munusamy, P.; Ng, M.J.; Sangrithi, M. Single-Cell RNA Sequencing of the Cynomolgus Macaque Testis Reveals Conserved Transcriptional Profiles during Mammalian Spermatogenesis. Dev. Cell 2020, 54, 548–566.e7. [Google Scholar] [CrossRef]
- Nabi, S.; Askari, M.; Rezaei-Gazik, M.; Salehi, N.; Almadani, N.; Tahamtani, Y.; Totonchi, M. A rare frameshift mutation in SYCP1 is associated with human male infertility. Mol. Hum. Reprod. 2022, 28, gaac009. [Google Scholar] [CrossRef]
- Billmyre, K.K.; Kesler, E.A.; Tsuchiya, D.; Corbin, T.J.; Weaver, K.; Moran, A.; Yu, Z.; Adams, L.; Delventhal, K.; Durnin, M.; et al. SYCP1 head-to-head assembly is required for chromosome synapsis in mouse meiosis. Sci. Adv. 2023, 9, eadi1562. [Google Scholar] [CrossRef]
- Ernst, C.; Eling, N.; Martinez-Jimenez, C.P.; Marioni, J.C.; Odom, D.T. Staged developmental mapping and X chromosome transcriptional dynamics during mouse spermatogenesis. Nat. Commun. 2019, 10, 1251. [Google Scholar] [CrossRef]
- Gonen, N.; Lovell-Badge, R. The regulation of Sox9 expression in the gonad. Curr. Top. Dev. Biol. 2019, 134, 223–252. [Google Scholar] [CrossRef]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2010, 107, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA–protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef]
- Lun, A.T.L.; Riesenfeld, S.; Andrews, T.; Dao, T.P.T.P.; Gomes, T.; Marioni, J.C. EmptyDrops: Distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 2019, 20, 63. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Sekine, K.; Gerber, T.; Loeffler-Wirth, H.; Binder, H.; Gac, M.; Kanton, S.; Kageyama, J.; Damm, G.; Seehofer, D.; et al. Multilineage communication regulates human liver bud development from pluripotency. Nature 2017, 546, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Z.; Wang, P.; Yang, Q.; Gun, S. Single-Cell RNA Sequencing Reveals an Atlas of Hezuo Pig Testis Cells. Int. J. Mol. Sci. 2024, 25, 9786. https://doi.org/10.3390/ijms25189786
Yan Z, Wang P, Yang Q, Gun S. Single-Cell RNA Sequencing Reveals an Atlas of Hezuo Pig Testis Cells. International Journal of Molecular Sciences. 2024; 25(18):9786. https://doi.org/10.3390/ijms25189786
Chicago/Turabian StyleYan, Zunqiang, Pengfei Wang, Qiaoli Yang, and Shuangbao Gun. 2024. "Single-Cell RNA Sequencing Reveals an Atlas of Hezuo Pig Testis Cells" International Journal of Molecular Sciences 25, no. 18: 9786. https://doi.org/10.3390/ijms25189786
APA StyleYan, Z., Wang, P., Yang, Q., & Gun, S. (2024). Single-Cell RNA Sequencing Reveals an Atlas of Hezuo Pig Testis Cells. International Journal of Molecular Sciences, 25(18), 9786. https://doi.org/10.3390/ijms25189786