
The NF-κB1/p50 Subunit Influences the Notch/IL-6-Driven Expansion of Myeloid-Derived Suppressor Cells in Murine T-Cell Acute Lymphoblastic Leukemia

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
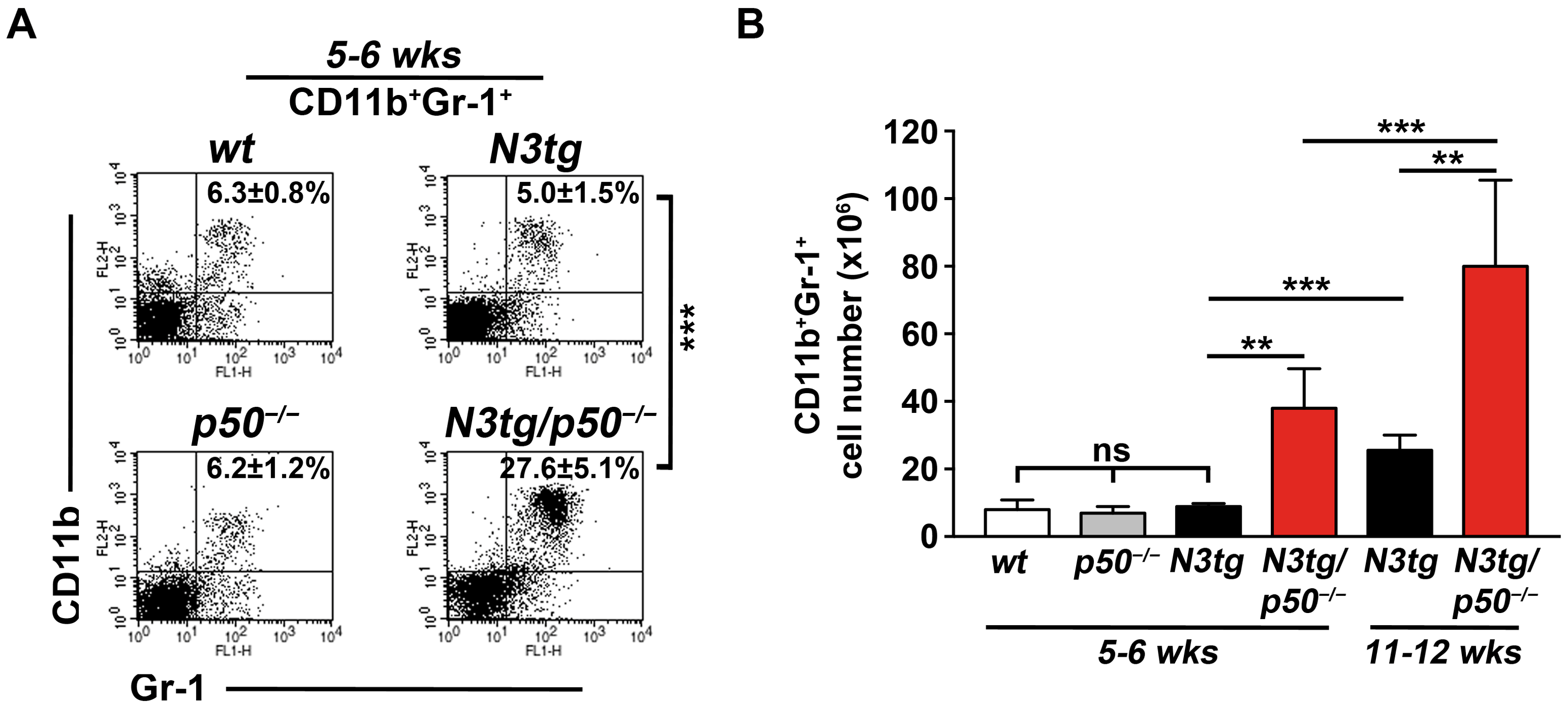
2.1. CD11b+GR-1+ Myeloid Cells Expand Significantly in N3tg/p50−/− Mice at an Initial Stage of the Disease
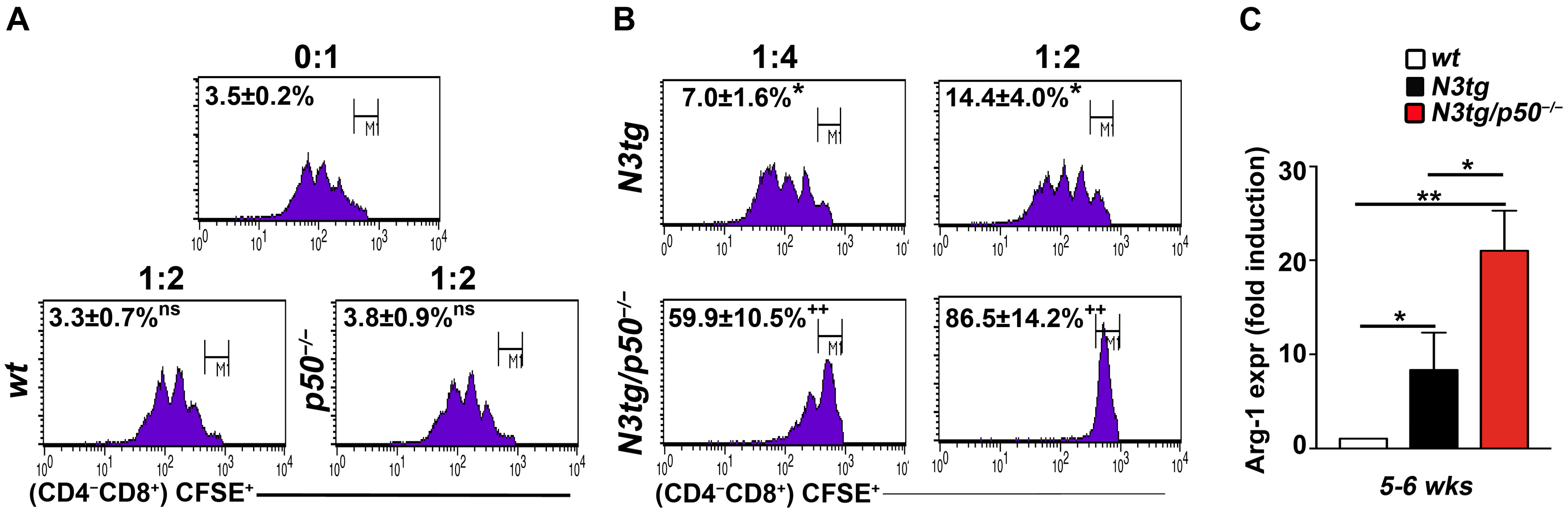
2.2. The Genetic Deletion of the NF-κB1/p50 Subunit in N3tg Mice Enhances the Suppressive Function of T-ALL-Induced CD11b+GR-1+ MDSCs
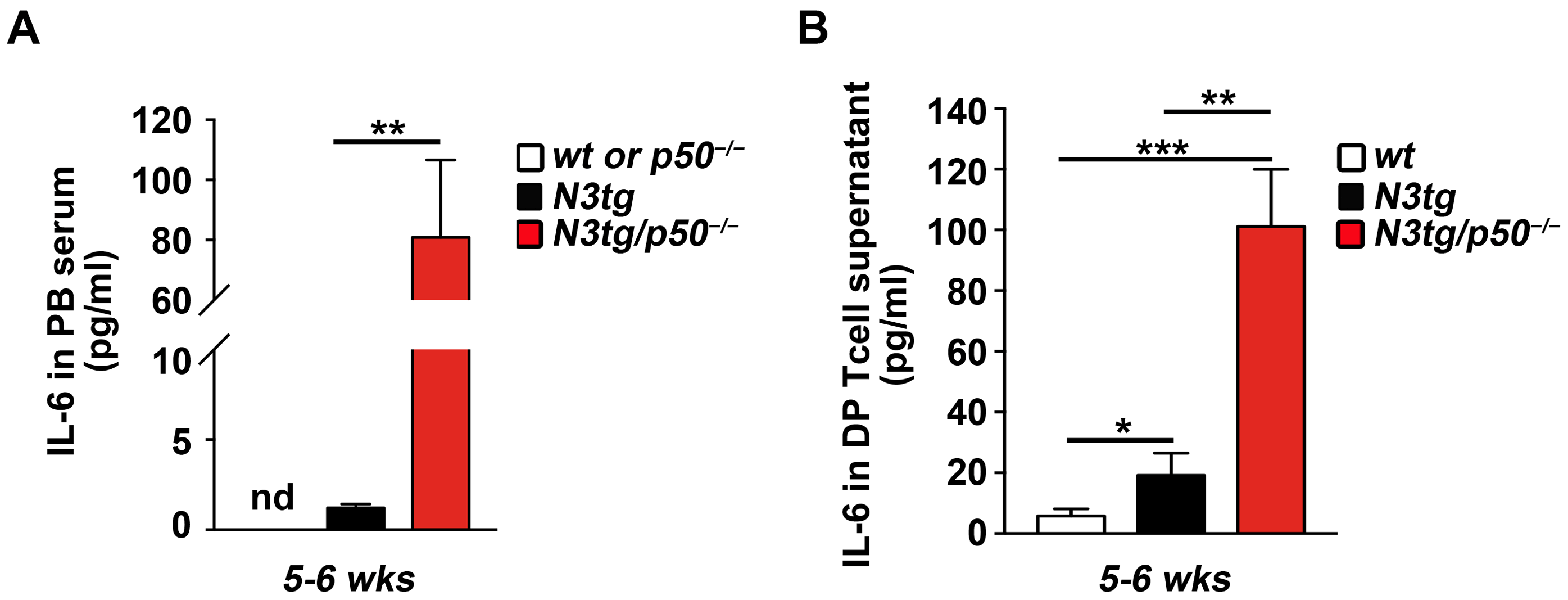
2.3. The Enhanced Expansion of CD11b+GR-1+ MDSCs in N3tg/p50−/− Double Mutant Mice Correlates with the Significant Increase in the IL-6 Cytokine
2.4. The Genetic Ablation of the NF-κB1/p50 Subunit Enhances the Transcription of the IL-6 Promoter in CD4+CD8+ DP T-Cells from N3tg/p50−/− Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cell Culture
4.3. Flow Cytometry and Cell Sorting
4.4. In Vitro Suppression Assay with Murine MDSCs
4.5. ELISA
4.6. Chromatin Immunoprecipitation Assay (ChIP-Assay)
- IL-6/NF-κB Site1: Fw 5′-TGGTAAATACAGAGCATTTGGGTG-3′;
- Rv 5′-TTGGGATAAAGTTGAGACAGGCT-3′;
- IL-6/NF-κB Site2: Fw 5′-AGCCATTGCCCCCAGGAT-3′;
- Rv 5′-GCACATATGTAGCAGAGGACTGT-3′;
- IL-6/NF-κB Site3: Fw 5′-CCTCTTCCCTGGGGTCTCA-3′;
- Rv 5′-TCAGAAGTCTCAACTAACCTGGAC-3′;
- IL-6/NF-κB Site4: Fw 5′-GGGGTTTCCAACTTCAGTCCA-3′;
- Rv 5′-AGTTGGTCCAATGACTAGCCC-3′.
4.7. RNA Isolation and RT-qPCR of Arginase-1
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shiraz, P.; Jehangir, W.; Agrawal, V. T-Cell Acute Lymphoblastic Leukemia-Current Concepts in Molecular Biology and Management. Biomedicines 2021, 9, 1621. [Google Scholar] [CrossRef] [PubMed]
- Gianni, F.; Belver, L.; Ferrando, A. The Genetics and Mechanisms of T-Cell Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2020, 10, a035246. [Google Scholar] [CrossRef] [PubMed]
- Cordo’, V.; van der Zwet, J.C.G.; Canté-Barrett, K.; Pieters, R.; Meijerink, J.P.P. T-cell Acute Lymphoblastic Leukemia: A Roadmap to Targeted Therapies. Blood Cancer Discov. 2020, 2, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Gao, X.; Wang, H. An Update on Clinical Trials and Potential Therapeutic Strategies in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2023, 24, 7201. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Mancuso, A.; Polerà, N.; Froio, C.; D’Aquino, G.; Riillo, C.; Tagliaferri, P.; Tassone, P. The emerging scenario of immunotherapy for T-cell Acute Lymphoblastic Leukemia: Advances, challenges and future perspectives. Exp. Hematol. Oncol. 2023, 12, 5. [Google Scholar] [CrossRef]
- Ho, D.M.; Guruharsha, K.G.; Artavanis-Tsakonas, S. The Notch Interactome: Complexity in Signaling Circuitry. Adv. Exp. Med. Biol. 2018, 1066, 125–140. [Google Scholar] [CrossRef]
- Shi, Q.; Xue, C.; Zeng, Y.; Yuan, X.; Chu, Q.; Jiang, S.; Wang, J.; Zhang, Y.; Zhu, D.; Li, L. Notch signaling pathway in cancer: From mechanistic insights to targeted therapies. Signal Transduct. Target. Ther. 2024, 9, 128. [Google Scholar] [CrossRef]
- Vanderbeck, A.; Maillard, I. Notch signaling at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2021, 109, 535–548. [Google Scholar] [CrossRef]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Ferreira, A.; Aster, J.C. Notch signaling in cancer: Complexity and challenges on the path to clinical translation. Semin. Cancer Biol. 2022, 85, 95–106. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P., 4th; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Bellavia, D.; Campese, A.F.; Vacca, A.; Gulino, A.; Screpanti, I. Notch3, another Notch in T cell development. Semin. Immunol. 2003, 15, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi-Elias, P.; Hu, T.; Jenkins, D.; Firestone, B.; Gans, S.; Kurth, E.; Capodieci, P.; Deplazes-Lauber, J.; Petropoulos, K.; Thiel, P.; et al. Characterization of activating mutations of NOTCH3 in T-cell acute lymphoblastic leukemia and anti-leukemic activity of NOTCH3 inhibitory antibodies. Oncogene 2016, 35, 6077–6086. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Severson, E.; Pear, W.S.; Liu, X.S.; Aster, J.C.; Blacklow, S.C. The common oncogenomic program of NOTCH1 and NOTCH3 signaling in T-cell acute lymphoblastic leukemia. PLoS ONE 2017, 12, e0185762. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; Helbig, C.; Backer, R.A. Notch in T Cell Differentiation: All Things Considered. Trends Immunol. 2015, 36, 802–814. [Google Scholar] [CrossRef]
- Chiplunkar, S.V.; Gogoi, D. The multifaceted role of Notch signal in regulating T cell fate. Immunol. Lett. 2019, 206, 59–64. [Google Scholar] [CrossRef]
- Brandstadter, J.D.; Maillard, I. Notch signalling in T cell homeostasis and differentiation. Open Biol. 2019, 9, 190187. [Google Scholar] [CrossRef]
- Shin, B.; Chang, S.J.; MacNabb, B.W.; Rothenberg, E.V. Transcriptional network dynamics in early T cell development. J. Exp. Med. 2024, 221, e20230893. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef]
- Toribio, M.L.; González-García, S. Notch Partners in the Long Journey of T-ALL Pathogenesis. Int. J. Mol. Sci. 2023, 24, 1383. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Pear, W.S.; Aster, J.C.; Scott, M.L.; Hasserjian, R.P.; Soffer, B.; Sklar, J.; Baltimore, D. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J. Exp. Med. 1996, 183, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, D.; Campese, A.F.; Alesse, E.; Vacca, A.; Felli, M.P.; Balestri, A.; Stoppacciaro, A.; Tiveron, C.; Tatangelo, L.; Giovarelli, M.; et al. Constitutive activation of NF-κB and T-cell leukemia/lymphoma in Notch3 transgenic mice. EMBO J. 2000, 19, 3337–3348. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.R.; Sanda, T.; Lawton, L.N.; Li, X.; Kreslavsky, T.; Novina, C.D.; Brand, M.; Gutierrez, A.; Kelliher, M.A.; Jamieson, C.H.; et al. The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J. Exp. Med. 2013, 210, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Wallaert, A.; Van Loocke, W.; Hernandez, L.; Taghon, T.; Speleman, F.; Van Vlierberghe, P. Comprehensive miRNA expression profiling in human T-cell acute lymphoblastic leukemia by small RNA-sequencing. Sci. Rep. 2017, 7, 7901. [Google Scholar] [CrossRef]
- Dawidowska, M.; Jaksik, R.; Drobna, M.; Szarzyńska-Zawadzka, B.; Kosmalska, M.; Sędek, Ł.; Machowska, L.; Lalik, A.; Lejman, M.; Ussowicz, M.; et al. Comprehensive Investigation of miRNome Identifies Novel Candidate miRNA-mRNA Interactions Implicated in T-Cell Acute Lymphoblastic Leukemia. Neoplasia 2019, 21, 294–310. [Google Scholar] [CrossRef]
- Del Gaizo, M.; Sergio, I.; Lazzari, S.; Cialfi, S.; Pelullo, M.; Screpanti, I.; Felli, M.P. MicroRNAs as Modulators of the Immune Response in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2022, 23, 829. [Google Scholar] [CrossRef]
- Sergio, I.; Varricchio, C.; Patel, S.K.; Del Gaizo, M.; Russo, E.; Orlando, A.; Peruzzi, G.; Ferrandino, F.; Tsaouli, G.; Coni, S.; et al. Notch3-regulated microRNAs impair CXCR4-dependent maturation of thymocytes allowing maintenance and progression of T-ALL. Oncogene 2024, 43, 2535–2547. [Google Scholar] [CrossRef]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef]
- Ferrandino, F.; Bernardini, G.; Tsaouli, G.; Grazioli, P.; Campese, A.F.; Noce, C.; Ciuffetta, A.; Vacca, A.; Besharat, Z.M.; Bellavia, D.; et al. Intrathymic Notch3 and CXCR4 combinatorial interplay facilitates T-cell leukemia propagation. Oncogene 2018, 37, 6285–6298. [Google Scholar] [CrossRef]
- Tsaouli, G.; Ferretti, E.; Bellavia, D.; Vacca, A.; Felli, M.P. Notch/CXCR4 Partnership in Acute Lymphoblastic Leukemia Progression. J. Immunol. Res. 2019, 2019, 5601396. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, R.; Cao, X.; Zhang, Y.; Sun, R.; Lu, W.; Zhao, M. Role of chemokines in T-cell acute lymphoblastic Leukemia: From pathogenesis to therapeutic options. Int. Immunopharmacol. 2023, 121, 110396. [Google Scholar] [CrossRef] [PubMed]
- Charbonnier, L.M.; Wang, S.; Georgiev, P.; Sefik, E.; Chatila, T.A. Control of peripheral tolerance by regulatory T cell-intrinsic Notch signaling. Nat. Immunol. 2015, 16, 1162–1173. [Google Scholar] [CrossRef]
- Grazioli, P.; Felli, M.P.; Screpanti, I.; Campese, A.F. The mazy case of Notch and immunoregulatory cells. J. Leukoc. Biol. 2017, 102, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Pastorczak, A.; Domka, K.; Fidyt, K.; Poprzeczko, M.; Firczuk, M. Mechanisms of Immune Evasion in Acute Lymphoblastic Leukemia. Cancers 2021, 13, 1536. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Lasser, S.A.; Ozbay Kurt, F.G.; Arkhypov, I.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat. Rev. Clin. Oncol. 2024, 21, 147–164. [Google Scholar] [CrossRef]
- Lu, J.; Luo, Y.; Rao, D.; Wang, T.; Lei, Z.; Chen, X.; Zhang, B.; Li, Y.; Liu, B.; Xia, L.; et al. Myeloid-derived suppressor cells in cancer: Therapeutic targets to overcome tumor immune evasion. Exp. Hematol. Oncol. 2024, 13, 39. [Google Scholar] [CrossRef]
- Fan, R.; De Beule, N.; Maes, A.; De Bruyne, E.; Menu, E.; Vanderkerken, K.; Maes, K.; Breckpot, K.; De Veirman, K. The prognostic value and therapeutic targeting of myeloid-derived suppressor cells in hematological cancers. Front. Immunol. 2022, 13, 1016059. [Google Scholar] [CrossRef]
- Grazioli, P.; Orlando, A.; Giordano, N.; Noce, C.; Peruzzi, G.; Abdollahzadeh, B.; Screpanti, I.; Campese, A.F. Notch-Signaling Deregulation Induces Myeloid-Derived Suppressor Cells in T-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2022, 13, 809261. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-κB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Betzler, A.C.; Theodoraki, M.N.; Schuler, P.J.; Döscher, J.; Laban, S.; Hoffmann, T.K.; Brunner, C. NF-κB and Its Role in Checkpoint Control. Int. J. Mol. Sci. 2020, 21, 3949. [Google Scholar] [CrossRef] [PubMed]
- Vilimas, T.; Mascarenhas, J.; Palomero, T.; Mandal, M.; Buonamici, S.; Meng, F.; Thompson, B.; Spaulding, C.; Macaroun, S.; Alegre, M.L.; et al. Targeting the NF-κB signaling pathway in Notch1-induced T-cell leukemia. Nat. Med. 2007, 13, 70–77. [Google Scholar] [CrossRef]
- Espinosa, L.; Cathelin, S.; D’Altri, T.; Trimarchi, T.; Statnikov, A.; Guiu, J.; Rodilla, V.; Inglés-Esteve, J.; Nomdedeu, J.; Bellosillo, B.; et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer Cell 2010, 18, 268–281. [Google Scholar] [CrossRef]
- Tsaouli, G.; Barbarulo, A.; Vacca, A.; Screpanti, I.; Felli, M.P. Molecular Mechanisms of Notch Signaling in Lymphoid Cell Lineages Development: NF-κB and Beyond. Adv. Exp. Med. Biol. 2020, 1227, 145–164. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Sig Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Grazioli, P.; Orlando, A.; Giordano, N.; Noce, C.; Peruzzi, G.; Scafetta, G.; Screpanti, I.; Campese, A.F. NF-κB1 Regulates Immune Environment and Outcome of Notch-Dependent T-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 11, 541. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef]
- Shin, H.M.; Minter, L.M.; Cho, O.H.; Gottipati, S.; Fauq, A.H.; Golde, T.E.; Sonenshein, G.E.; Osborne, B.A. Notch1 augments NF-κB activity by facilitating its nuclear retention. EMBO J. 2006, 25, 129–138. [Google Scholar] [CrossRef] [PubMed]
- de Valle, E.; Grigoriadis, G.; O’Reilly, L.A.; Willis, S.N.; Maxwell, M.J.; Corcoran, L.M.; Tsantikos, E.; Cornish, J.K.; Fairfax, K.A.; Vasanthakumar, A.; et al. NFκB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J. Exp. Med. 2016, 213, 621–641. [Google Scholar] [CrossRef]
- Wang, M.; Yu, F.; Zhang, Y.; Li, P. Novel insights into Notch signaling in tumor immunity: Potential targets for cancer immunotherapy. Front. Immunol. 2024, 15, 1352484. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Consonni, F.M.; Morlacchi, S.; Sangaletti, S.; Bleve, A.; Totaro, M.G.; Larghi, P.; Rimoldi, M.; Tripodo, C.; Strauss, L.; et al. Tumor-Derived Prostaglandin E2 Promotes p50 NF-κB-Dependent Differentiation of Monocytic MDSCs. Cancer Res. 2020, 80, 2874–2888. [Google Scholar] [CrossRef]
- Sangaletti, S.; Talarico, G.; Chiodoni, C.; Cappetti, B.; Botti, L.; Portararo, P.; Gulino, A.; Consonni, F.M.; Sica, A.; Randon, G.; et al. SPARC Is a New Myeloid-Derived Suppressor Cell Marker Licensing Suppressive Activities. Front. Immunol. 2019, 10, 1369. [Google Scholar] [CrossRef]
- Wang, L.; Dou, X.; Chen, S.; Yu, X.; Huang, X.; Zhang, L.; Chen, Y.; Wang, J.; Yang, K.; Bugno, J.; et al. YTHDF2 inhibition potentiates radiotherapy antitumor efficacy. Cancer Cell 2023, 41, 1294–1308. [Google Scholar] [CrossRef] [PubMed]
- Lalle, G.; Twardowski, J.; Grinberg-Bleyer, Y. NF-κB in Cancer Immunity: Friend or Foe? Cells 2021, 10, 355. [Google Scholar] [CrossRef]
- Weber, R.; Groth, C.; Lasser, S.; Arkhypov, I.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell. Immunol. 2021, 359, 104254. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef]
- Libermann, T.A.; Baltimore, D. Activation of interleukin-6 gene expression through the NF-κB transcription factor. Mol. Cell. Biol. 1990, 10, 2327–2334. [Google Scholar] [CrossRef]
- Miyazawa, K.; Mori, A.; Yamamoto, K.; Okudaira, H. Transcriptional roles of CCAAT/enhancer binding protein-β, nuclear factor-κB, and C-promoter binding factor 1 in interleukin (IL)-1β-induced IL-6 synthesis by human rheumatoid fibroblast-like synoviocytes. J. Biol. Chem. 1998, 273, 7620–7627. [Google Scholar] [CrossRef] [PubMed]
- Wongchana, W.; Palaga, T. Direct regulation of interleukin-6 expression by Notch signaling in macrophages. Cell. Mol. Immunol. 2012, 9, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lewander, A.; Butchi, A.K.; Gao, J.; He, L.J.; Lindblom, A.; Arbman, G.; Carstensen, J.; Zhang, Z.Y.; Sun, X.F. Swedish Low-Risk Colorectal Cancer Study Group. Polymorphism in the promoter region of the NFKB1 gene increases the risk of sporadic colorectal cancer in Swedish but not in Chinese populations. Scand. J. Gastroenterol. 2007, 42, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, C.; Cai, Y.; Liu, B. Association of NFKB1 and NFKBIA gene polymorphisms with susceptibility of gastric cancer. Tumour Biol. 2017, 39, 1010428317717107. [Google Scholar] [CrossRef] [PubMed]
- Karban, A.S.; Okazaki, T.; Panhuysen, C.I.; Gallegos, T.; Potter, J.J.; Bailey-Wilson, J.E.; Silverberg, M.S.; Duerr, R.H.; Cho, J.H.; Gregersen, P.K.; et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum. Mol. Genet. 2004, 13, 35–45. [Google Scholar] [CrossRef]
- Giachelia, M.; Voso, M.T.; Tisi, M.C.; Martini, M.; Bozzoli, V.; Massini, G.; D’Aló, F.; Larocca, L.M.; Leone, G.; Hohaus, S. Interleukin-6 plasma levels are modulated by a polymorphism in the NF-κB1 gene and are associated with outcome following rituximab-combined chemotherapy in diffuse large B-cell non-Hodgkin lymphoma. Leuk. Lymphoma 2012, 53, 411–416. [Google Scholar] [CrossRef]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef]
- Calzado, M.A.; Bacher, S.; Schmitz, M.L. NF-κB inhibitors for the treatment of inflammatory diseases and cancer. Curr. Med. Chem. 2007, 14, 367–376. [Google Scholar] [CrossRef]
- Paul, A.; Edwards, J.; Pepper, C.; Mackay, S. Inhibitory-κB Kinase (IKK) α and Nuclear Factor-κB (NFκB)-Inducing Kinase (NIK) as Anti-Cancer Drug Targets. Cells 2018, 7, 176. [Google Scholar] [CrossRef]
- Prescott, J.A.; Cook, S.J. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Sha, W.C.; Liou, H.C.; Tuomanen, E.I.; Baltimore, D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell 1995, 80, 321–330. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdollahzadeh, B.; Cantale Aeo, N.M.; Giordano, N.; Orlando, A.; Basciani, M.; Peruzzi, G.; Grazioli, P.; Screpanti, I.; Felli, M.P.; Campese, A.F. The NF-κB1/p50 Subunit Influences the Notch/IL-6-Driven Expansion of Myeloid-Derived Suppressor Cells in Murine T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2024, 25, 9882. https://doi.org/10.3390/ijms25189882
Abdollahzadeh B, Cantale Aeo NM, Giordano N, Orlando A, Basciani M, Peruzzi G, Grazioli P, Screpanti I, Felli MP, Campese AF. The NF-κB1/p50 Subunit Influences the Notch/IL-6-Driven Expansion of Myeloid-Derived Suppressor Cells in Murine T-Cell Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2024; 25(18):9882. https://doi.org/10.3390/ijms25189882
Chicago/Turabian StyleAbdollahzadeh, Behnaz, Noemi Martina Cantale Aeo, Nike Giordano, Andrea Orlando, Maria Basciani, Giovanna Peruzzi, Paola Grazioli, Isabella Screpanti, Maria Pia Felli, and Antonio Francesco Campese. 2024. "The NF-κB1/p50 Subunit Influences the Notch/IL-6-Driven Expansion of Myeloid-Derived Suppressor Cells in Murine T-Cell Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 25, no. 18: 9882. https://doi.org/10.3390/ijms25189882
APA StyleAbdollahzadeh, B., Cantale Aeo, N. M., Giordano, N., Orlando, A., Basciani, M., Peruzzi, G., Grazioli, P., Screpanti, I., Felli, M. P., & Campese, A. F. (2024). The NF-κB1/p50 Subunit Influences the Notch/IL-6-Driven Expansion of Myeloid-Derived Suppressor Cells in Murine T-Cell Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences, 25(18), 9882. https://doi.org/10.3390/ijms25189882