
Reduction of Mitochondrial Calcium Overload via MKT077-Induced Inhibition of Glucose-Regulated Protein 75 Alleviates Skeletal Muscle Pathology in Dystrophin-Deficient mdx Mice

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
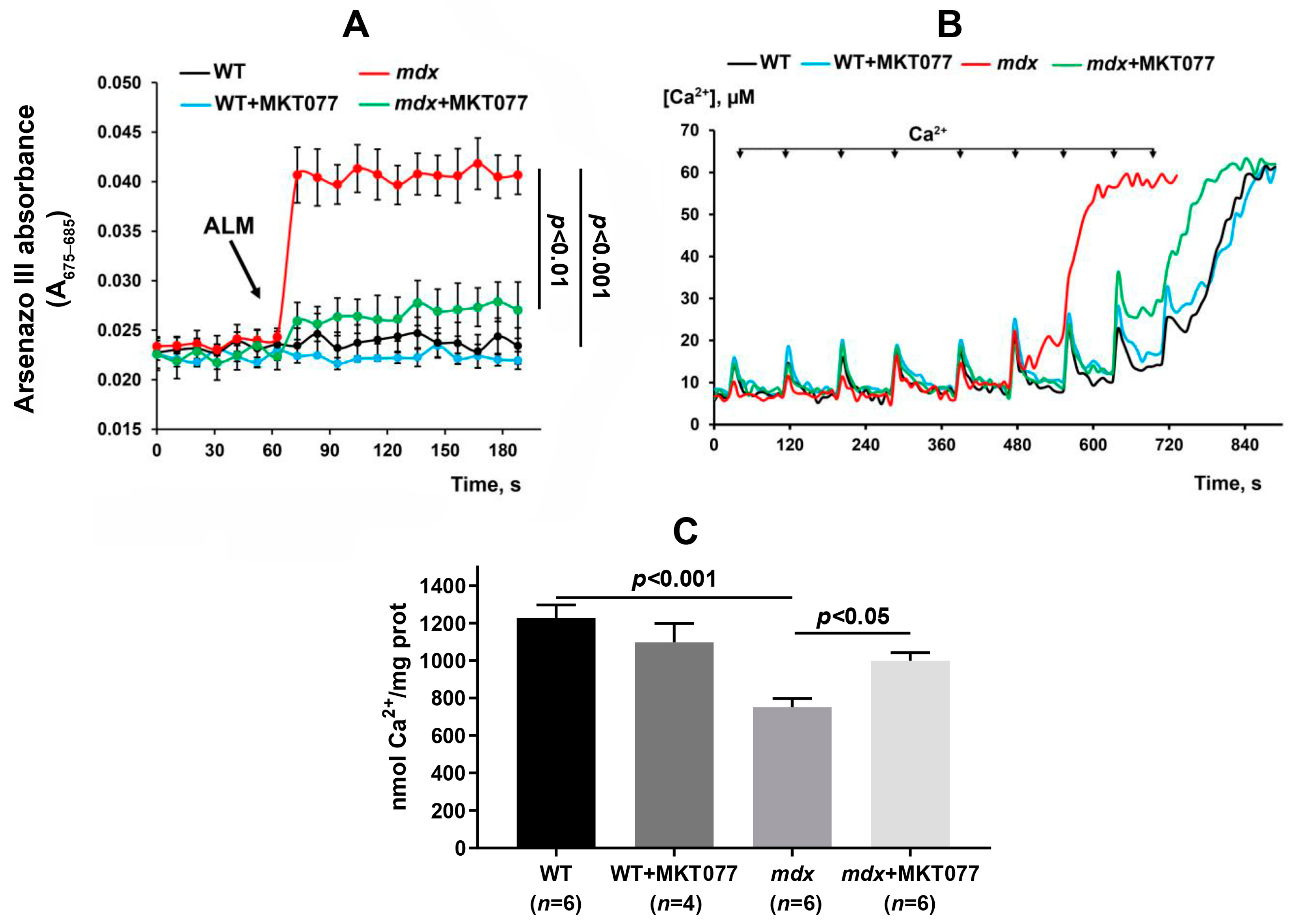
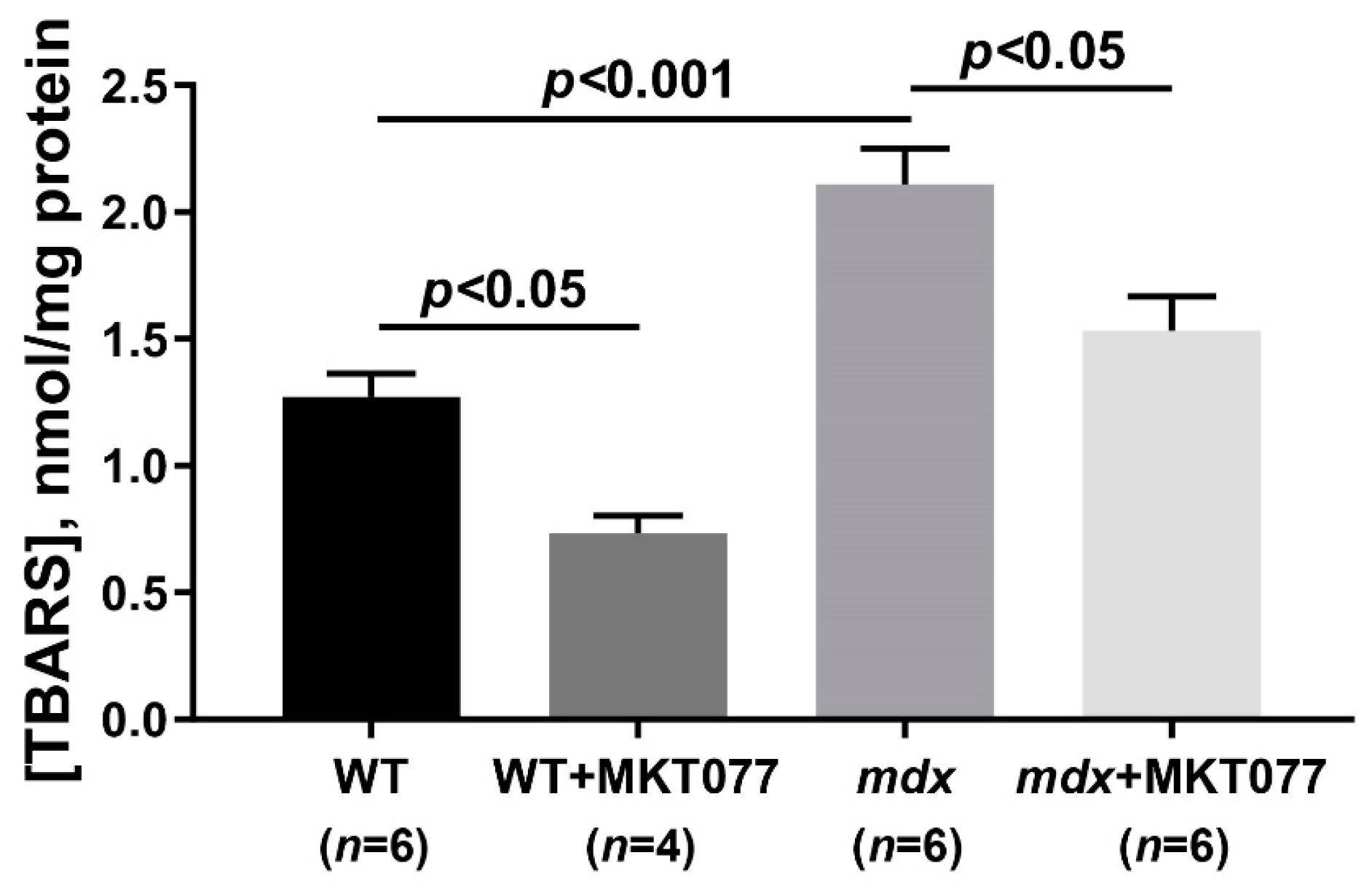
2.1. MKT077 Reduces Calcium Overload and Oxidative Stress in Skeletal Muscle Mitochondria of mdx Mice but Has No Effect on Impaired Oxidative Phosphorylation and Reduced ATP Levels
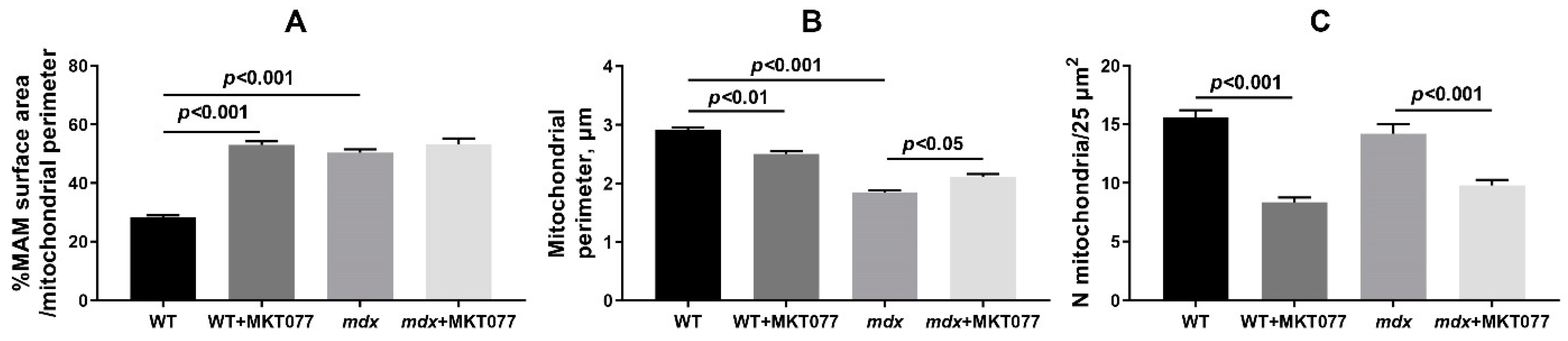
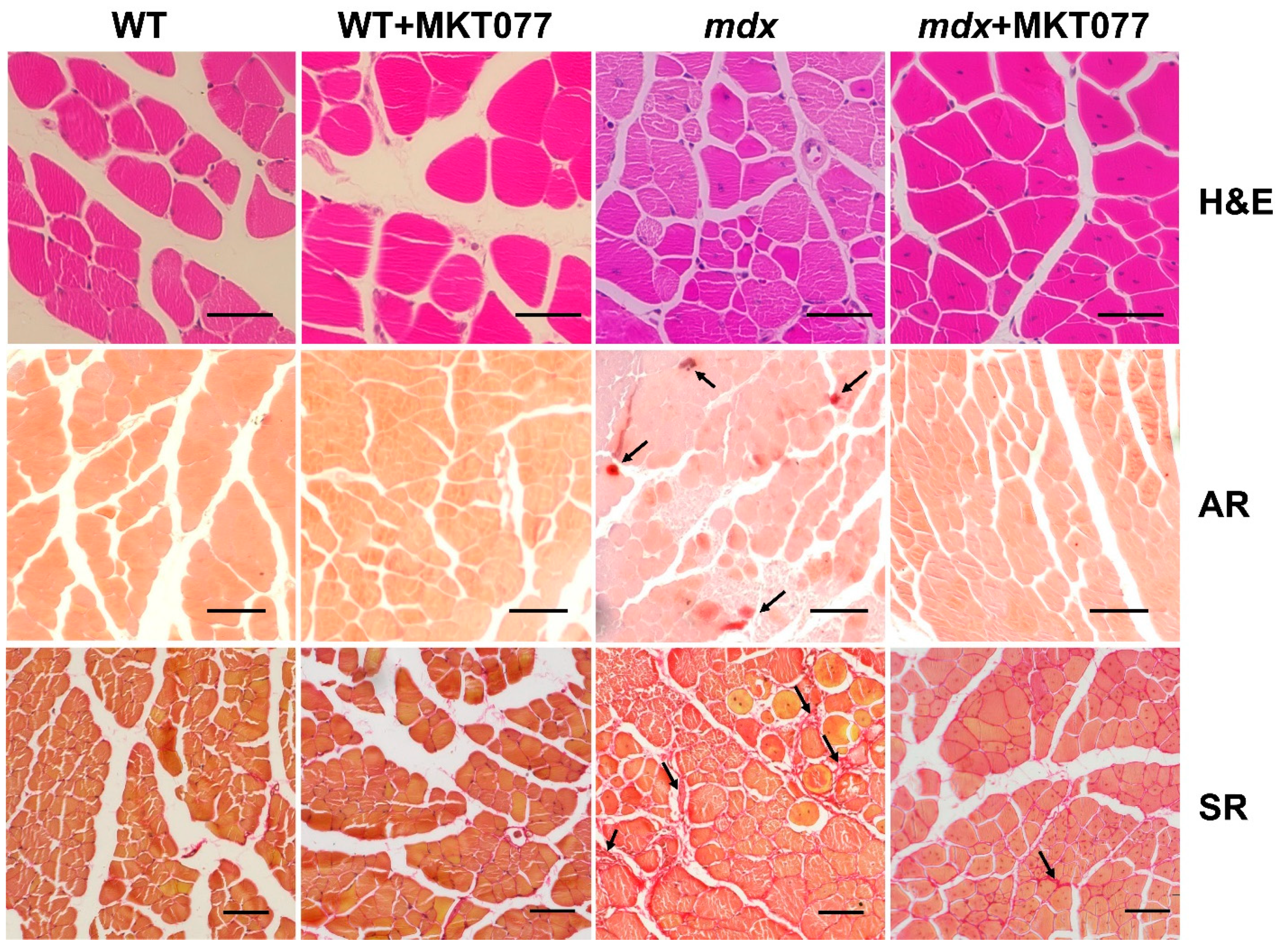
2.2. MKT077 Improves Skeletal Muscle Ultrastructure in mdx Mice but Causes Impairments in WT Animals
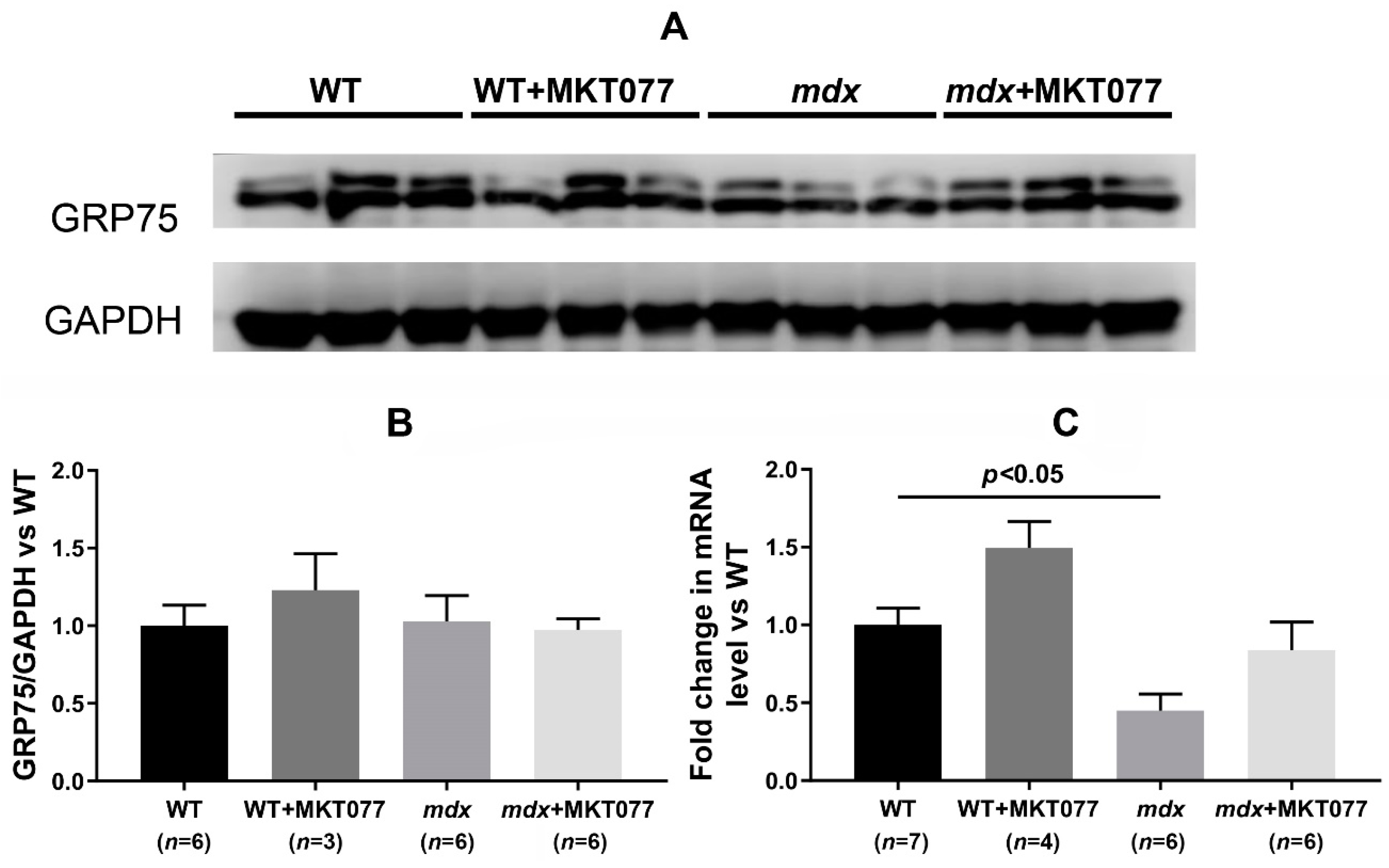
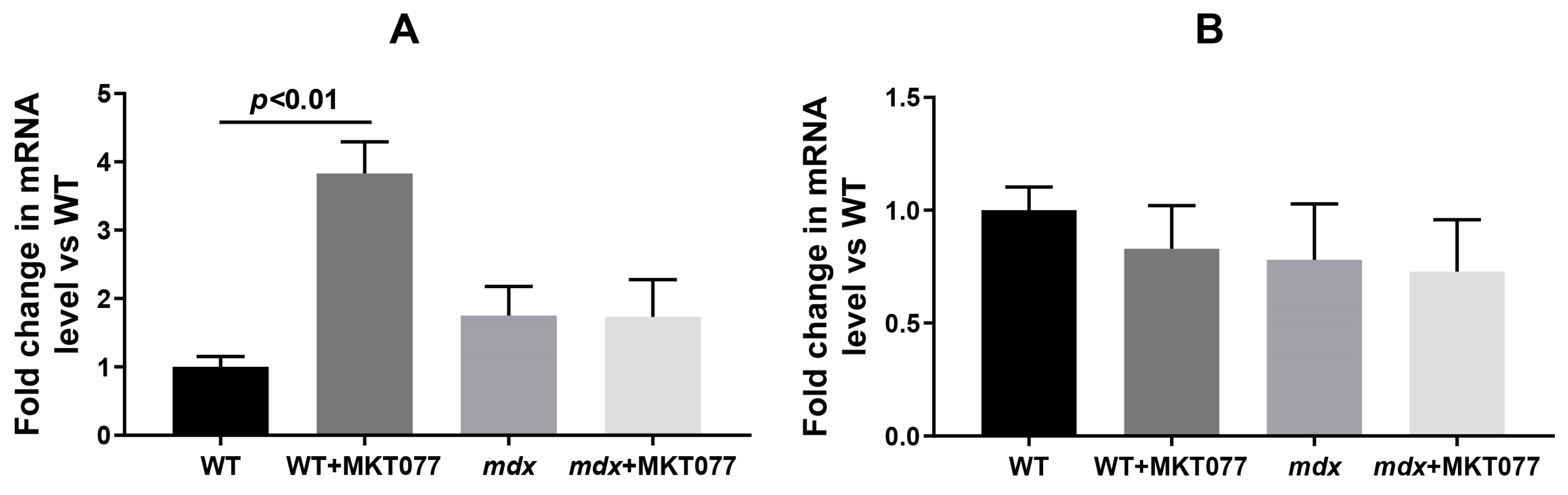
2.3. MKT077 Has No Effect on Increased SR Stress in Quadriceps of mdx Mice but Increases SR Stress in Muscles of WT Mice
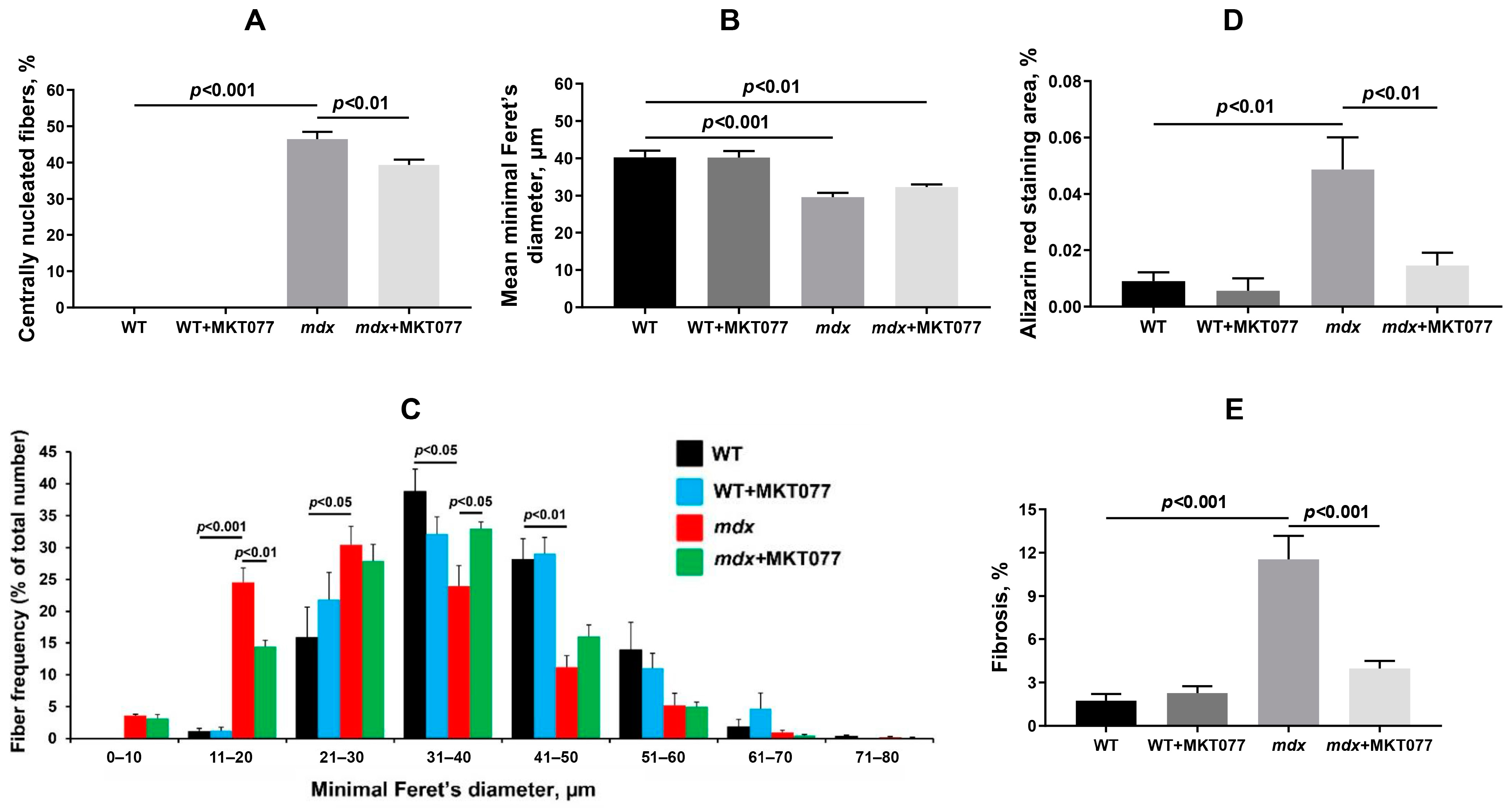
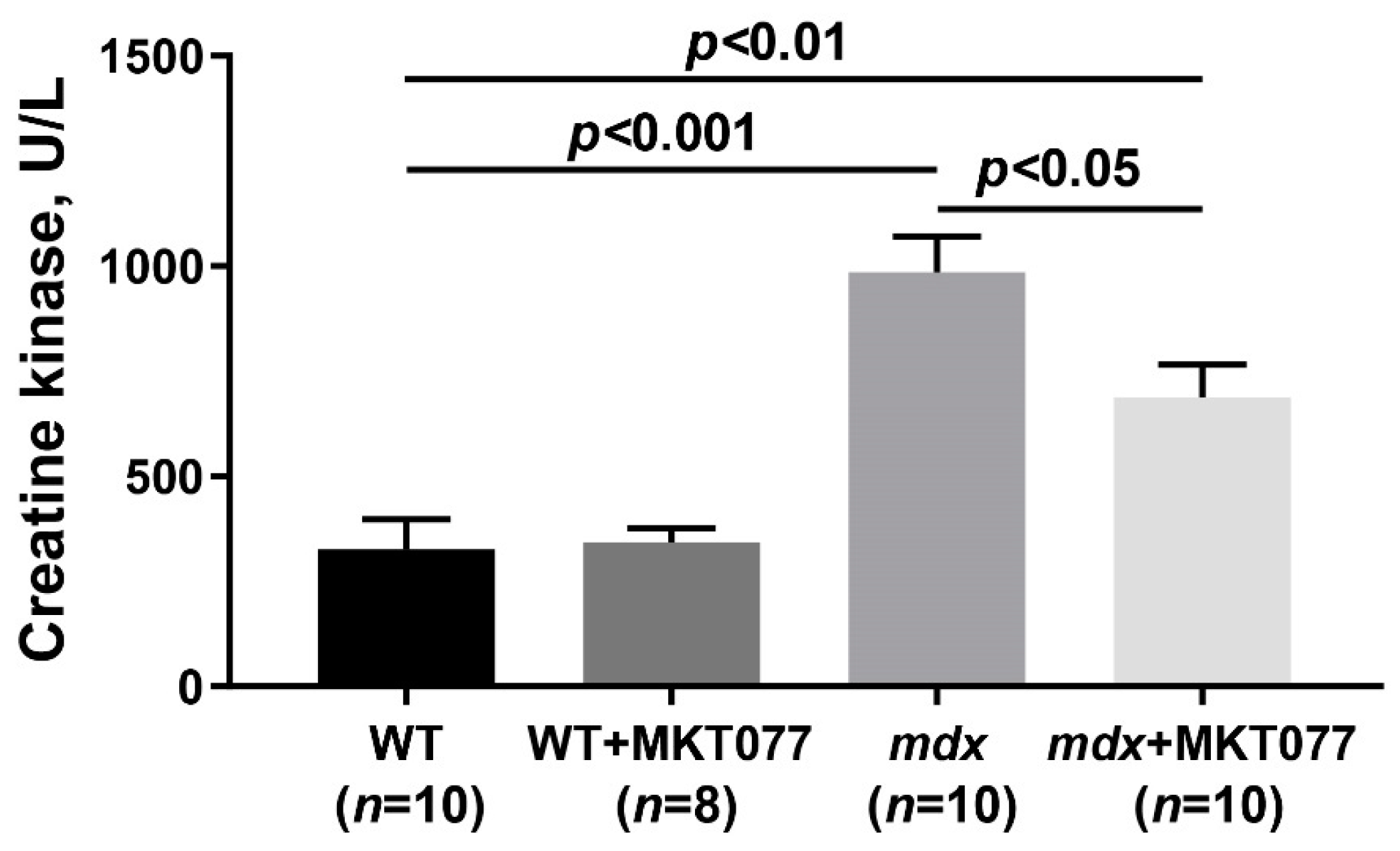
2.4. MKT077 Reduces Skeletal Muscle Degeneration and Enhances Grip Strength of mdx Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Grip Strength and Wire-Hanging Tests
4.3. Creatine Kinase Assay
4.4. Transmission Electron Microscopy
4.5. Histological Analysis
4.6. Quantitative Real-Time PCR
4.7. Electrophoresis and Western Blotting
4.8. Isolation of Skeletal Muscle Mitochondria and Their Functional Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monaco, A.P.; Neve, R.L.; Colletti-Feener, C.; Bertelson, C.J.; Kurnit, D.M.; Kunkel, L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Belosludtsev, K.N. Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy. Int. J. Mol. Sci. 2023, 24, 2229. [Google Scholar] [CrossRef]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front. Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef]
- Crescenzo, R.; Bianco, F.; Mazzoli, A.; Giacco, A.; Liverini, G.; Iossa, S. Skeletal Muscle Mitochondrial Energetic Efficiency and Aging. Int. J. Mol. Sci. 2015, 16, 10674–10685. [Google Scholar] [CrossRef]
- Gellerich, F.N.; Gizatullina, Z.; Trumbeckaite, S.; Nguyen, H.P.; Pallas, T.; Arandarcikaite, O.; Vielhaber, S.; Seppet, E.; Striggow, F. The regulation of OXPHOS by extramitochondrial calcium. Biochim. Biophys. Acta 2010, 1797, 1018–1027. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Mikheeva, I.B.; Stepanova, A.E.; Igoshkina, A.D.; Cherepanova, A.A.; Semenova, A.A.; Sharapov, V.A.; Kireev, I.I.; Belosludtsev, K.N. Mitochondrial Transplantation Therapy Ameliorates Muscular Dystrophy in mdx Mouse Model. Biomolecules 2024, 14, 316. [Google Scholar] [CrossRef]
- Bround, M.J.; Havens, J.R.; York, A.J.; Sargent, M.A.; Karch, J.; Molkentin, J.D. ANT-dependent MPTP underlies necrotic myofiber death in muscular dystrophy. Sci. Adv. 2023, 9, eadi2767. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K.N. Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef]
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. 2010, 20, 753–760. [Google Scholar] [CrossRef]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Šileikytė, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef] [PubMed]
- Simoes, I.C.M.; Morciano, G.; Lebiedzinska-Arciszewska, M.; Aguiari, G.; Pinton, P.; Potes, Y.; Wieckowski, M.R. The mystery of mitochondria-ER contact sites in physiology and pathology: A cancer perspective. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165834. [Google Scholar] [CrossRef]
- Yuan, M.; Gong, M.; He, J.; Xie, B.; Zhang, Z.; Meng, L.; Tse, G.; Zhao, Y.; Bao, Q.; Zhang, Y.; et al. IP3R1/GRP75/VDAC1 complex mediates endoplasmic reticulum stress-mitochondrial oxidative stress in diabetic atrial remodeling. Redox Biol. 2022, 52, 102289. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.Y.; Shao, J.; Zhu, L.; Xie, T.H.; Cai, J.; Wang, W.; Cai, M.X.; Wang, Z.L.; Yao, Y.; et al. GRP75 Modulates Endoplasmic Reticulum-Mitochondria Coupling and Accelerates Ca2+-Dependent Endothelial Cell Apoptosis in Diabetic Retinopathy. Biomolecules 2022, 12, 1778. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.Y.; Bu, D.F.; Zhang, Y.; Zhu, S.N. IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, Q.; Cao, B.; Cai, Y.; Wen, S.; Bian, J.; Zou, H.; Song, R.; Gu, J.; Liu, X.; et al. Ca2+ transfer via the ER-mitochondria tethering complex in neuronal cells contribute to cadmium-induced autophagy. Cell Biol. Toxicol. 2022, 38, 469–485. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, B.; Sheng, J.; Wang, J.; Zhu, W.; Xie, C.; Zhou, X.; Zhang, Y.; Meng, Q.; Li, Y. Potential targets for the treatment of MI: GRP75-mediated Ca2+ transfer in MAM. Eur. J. Pharmacol. 2024, 971, 176530. [Google Scholar] [CrossRef] [PubMed]
- Rousaki, A.; Miyata, Y.; Jinwal, U.K.; Dickey, C.A.; Gestwicki, J.E.; Zuiderweg, E.R. Allosteric drugs: The interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J. Mol. Biol. 2011, 411, 614–632. [Google Scholar] [CrossRef]
- Wen, B.; Xu, K.; Huang, R.; Jiang, T.; Wang, J.; Chen, J.; Chen, J.; He, B. Preserving mitochondrial function by inhibiting GRP75 ameliorates neuron injury under ischemic stroke. Mol. Med. Rep. 2022, 25, 165. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Starinets, V.S.; Belosludtseva, N.V.; Mikheeva, I.B.; Chelyadnikova, Y.A.; Igoshkina, A.D.; Vafina, A.B.; Vedernikov, A.A.; Belosludtsev, K.N. BKCa Activator NS1619 Improves the Structure and Function of Skeletal Muscle Mitochondria in Duchenne Dystrophy. Pharmaceutics 2022, 14, 2336. [Google Scholar] [CrossRef]
- Bellissimo, C.A.; Garibotti, M.C.; Perry, C.G.R. Mitochondrial stress responses in Duchenne muscular dystrophy: Metabolic dysfunction or adaptive reprogramming? Am. J. Physiol. Cell Physiol. 2022, 323, C718–C730. [Google Scholar] [CrossRef] [PubMed]
- Pauly, M.; Angebault-Prouteau, C.; Dridi, H.; Notarnicola, C.; Scheuermann, V.; Lacampagne, A.; Matecki, S.; Fauconnier, J. ER stress disturbs SR/ER-mitochondria Ca2+ transfer: Implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 2229–2239. [Google Scholar] [CrossRef]
- Romero, N.B.; Bitoun, M. Centronuclear myopathies. Semin. Pediatr. Neurol. 2011, 18, 250–256. [Google Scholar] [CrossRef]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef]
- Young, C.N.J.; Gosselin, M.R.F.; Rumney, R.; Oksiejuk, A.; Chira, N.; Bozycki, L.; Matryba, P.; Lukasiewicz, K.; Kao, A.P.; Dunlop, J.; et al. Total Absence of Dystrophin Expression Exacerbates Ectopic Myofiber Calcification and Fibrosis and Alters Macrophage Infiltration Patterns. Am. J. Pathol. 2020, 190, 190–205. [Google Scholar] [CrossRef]
- Houang, E.M.; Haman, K.J.; Filareto, A.; Perlingeiro, R.C.; Bate, F.S.; Lowe, D.A.; Metzger, J.M. Membrane-stabilizing copolymers confer marked protection to dystrophic skeletal muscle in vivo. Mol. Ther.-Methods Clin. Dev. 2015, 2, 15042. [Google Scholar] [CrossRef]
- Matsumura, C.Y.; Pertille, A.; Albuquerque, T.C.; Santo Neto, H.; Marques, M.J. Diltiazem and verapamil protect dystrophin-deficient muscle fibers of MDX mice from degeneration: A potential role in calcium buffering and sarcolemmal stability. Muscle Nerve 2009, 39, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, F.; Valladares, D.; Henríquez-Olguín, C.; Casas, M.; López, J.R.; Allen, P.D.; Jaimovich, E. Nifedipine treatment reduces resting calcium concentration, oxidative and apoptotic gene expression, and improves muscle function in dystrophic mdx mice. PLoS ONE 2013, 8, e81222. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Koya, K.; Weisberg, E.; Brunelli, B.T.; Li, Y.; Chen, L.B. Selective damage to carcinoma mitochondria by the rhodacyanine MKT-077. Cancer Res. 1996, 56, 544–550. [Google Scholar] [PubMed]
- Weisberg, E.L.; Koya, K.; Modica-Napolitano, J.; Li, Y.; Chen, L.B. In vivo administration of MKT-077 causes partial yet reversible impairment of mitochondrial function. Cancer Res. 1996, 56, 551–555. [Google Scholar] [PubMed]
- Liu, Y.; Jin, M.; Wang, Y.; Zhu, J.; Tan, R.; Zhao, J.; Ji, X.; Jin, C.; Jia, Y.; Ren, T.; et al. MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal Transduct. Target. Ther. 2020, 5, 59. [Google Scholar] [CrossRef]
- Cook, K.L.; Soto-Pantoja, D.R.; Abu-Asab, M.; Clarke, P.A.; Roberts, D.D.; Clarke, R. Mitochondria directly donate their membrane to form autophagosomes during a novel mechanism of parkin-associated mitophagy. Cell Biosci. 2014, 4, 16. [Google Scholar] [CrossRef]
- Abunimer, A.N.; Mohammed, H.; Cook, K.L.; Soto-Pantoja, D.R.; Campos, M.M.; Abu-Asab, M.S. Mitochondrial autophagosomes as a mechanism of drug resistance in breast carcinoma. Ultrastruct. Pathol. 2018, 42, 170–180. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, N.; Wu, B.; Wu, S.; Zhang, Y.; Sun, Y. The Role of Mitochondria in Vascular Calcification. J. Transl. Intern. Med. 2020, 8, 80–90. [Google Scholar] [CrossRef]
- Kumar, A.; Accorsi, A.; Rhee, Y.; Girgenrath, M. Do’s and don’ts in the preparation of muscle cryosections for histological analysis. J. Vis. Exp. 2015, 99, e52793. [Google Scholar] [CrossRef]
- Thoudam, T.; Ha, C.M.; Leem, J.; Chanda, D.; Park, J.S.; Kim, H.J.; Jeon, J.H.; Choi, Y.K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 Augments ER-Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Dragon, A.H.; Rowe, C.J.; Rhodes, A.M.; Pak, O.L.; Davis, T.A.; Ronzier, E. Systematic Identification of the Optimal Housekeeping Genes for Accurate Transcriptomic and Proteomic Profiling of Tissues following Complex Traumatic Injury. Methods Protoc. 2023, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. J. Biol. Chem. 1955, 217, 383–393. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | V Respiration, nmol O2/min per 1 mg of Protein | RCR (Relative Units) | |||
|---|---|---|---|---|---|
| State 2 | State 3 | State 4 | State 3UDNP | ||
| WT (n = 6) | 32.8 ± 1.9 | 204.0 ± 3.0 | 33.3 ± 2.2 | 276.7 ± 8.6 | 6.0 ± 0.2 |
| WT+MKT077 (n = 4) | 31.1 ± 1.8 | 159.5 ± 6.8 * | 33.3 ± 2.2 | 209.7 ± 11.4 * | 4.8 ± 0.2 * |
| mdx (n = 6) | 32.2 ± 2.1 | 175.0 ± 7.2 * | 35.8 ± 2.5 | 229.5 ± 13.5 * | 5.0 ± 0.2 * |
| mdx+MKT077 (n = 6) | 28.8 ± 2.8 | 156.2 ± 9.1 * | 32.7 ± 3.3 | 210.6 ± 10.9 * | 4.9 ± 0.2 * |
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Hspa5 | TGAAGAGCTGAACATGGACC | CTCATCGGGGTTTATGCCAC |
| Hspa9 | GACAAGGATGCCCAAGGTTC | GTAAAGACGCCTCCCAGAGT |
| Pink1 | TTGCCCCACACCCTAACATC | GCAGGGTACAGGGGTAGTTCT |
| Parkin | AGCCAGAGGTCCAGCAGTTA | GAGGGTTGCTTGTTTGCAGG |
| Rplp2 | CGGCTCAACAAGGTCATCAGTGA | AGCAGAAACAGCCACAGCCCCAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubinin, M.V.; Stepanova, A.E.; Mikheeva, I.B.; Igoshkina, A.D.; Cherepanova, A.A.; Talanov, E.Y.; Khoroshavina, E.I.; Belosludtsev, K.N. Reduction of Mitochondrial Calcium Overload via MKT077-Induced Inhibition of Glucose-Regulated Protein 75 Alleviates Skeletal Muscle Pathology in Dystrophin-Deficient mdx Mice. Int. J. Mol. Sci. 2024, 25, 9892. https://doi.org/10.3390/ijms25189892
Dubinin MV, Stepanova AE, Mikheeva IB, Igoshkina AD, Cherepanova AA, Talanov EY, Khoroshavina EI, Belosludtsev KN. Reduction of Mitochondrial Calcium Overload via MKT077-Induced Inhibition of Glucose-Regulated Protein 75 Alleviates Skeletal Muscle Pathology in Dystrophin-Deficient mdx Mice. International Journal of Molecular Sciences. 2024; 25(18):9892. https://doi.org/10.3390/ijms25189892
Chicago/Turabian StyleDubinin, Mikhail V., Anastasia E. Stepanova, Irina B. Mikheeva, Anastasia D. Igoshkina, Alena A. Cherepanova, Eugeny Yu. Talanov, Ekaterina I. Khoroshavina, and Konstantin N. Belosludtsev. 2024. "Reduction of Mitochondrial Calcium Overload via MKT077-Induced Inhibition of Glucose-Regulated Protein 75 Alleviates Skeletal Muscle Pathology in Dystrophin-Deficient mdx Mice" International Journal of Molecular Sciences 25, no. 18: 9892. https://doi.org/10.3390/ijms25189892
APA StyleDubinin, M. V., Stepanova, A. E., Mikheeva, I. B., Igoshkina, A. D., Cherepanova, A. A., Talanov, E. Y., Khoroshavina, E. I., & Belosludtsev, K. N. (2024). Reduction of Mitochondrial Calcium Overload via MKT077-Induced Inhibition of Glucose-Regulated Protein 75 Alleviates Skeletal Muscle Pathology in Dystrophin-Deficient mdx Mice. International Journal of Molecular Sciences, 25(18), 9892. https://doi.org/10.3390/ijms25189892