
Disulfide Bond Engineering of Soluble ACE2 for Thermal Stability Enhancement

Abstract
:1. Introduction
2. Results and Discussion
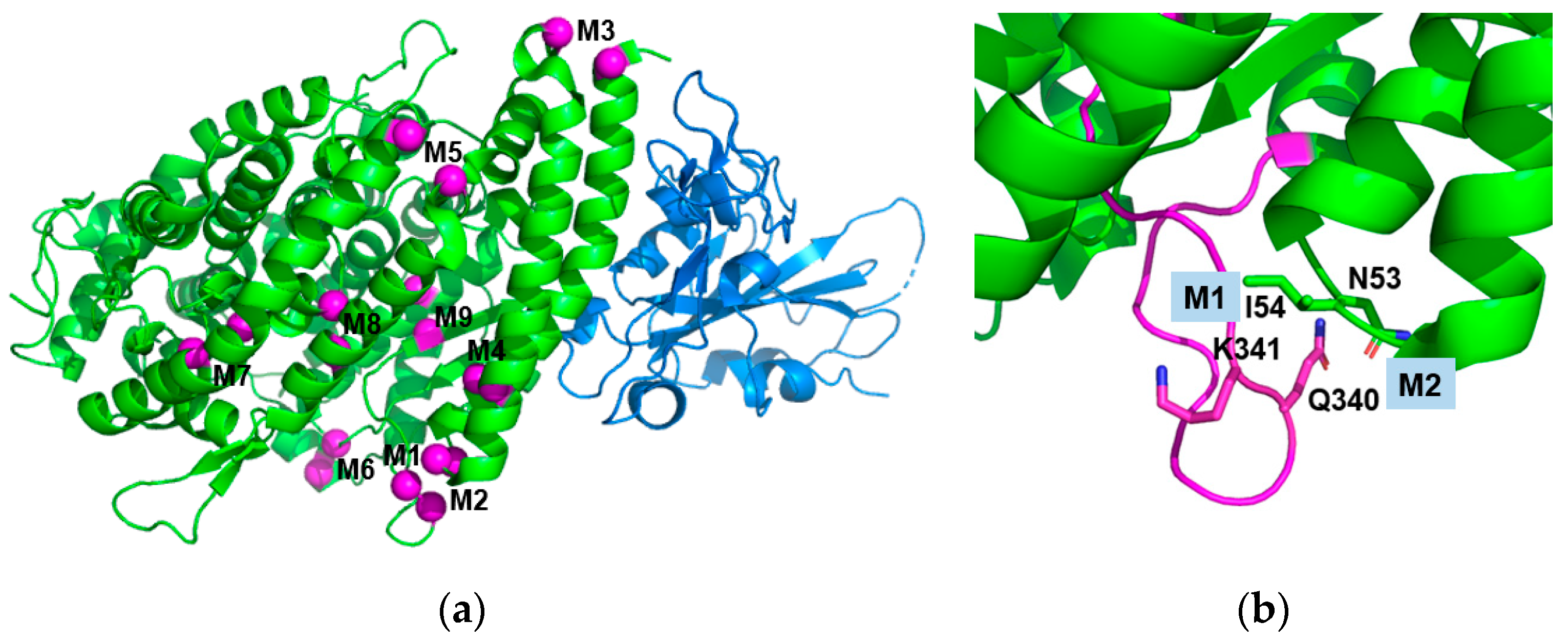
2.1. Design of Stabilizing Disulfide Bonds
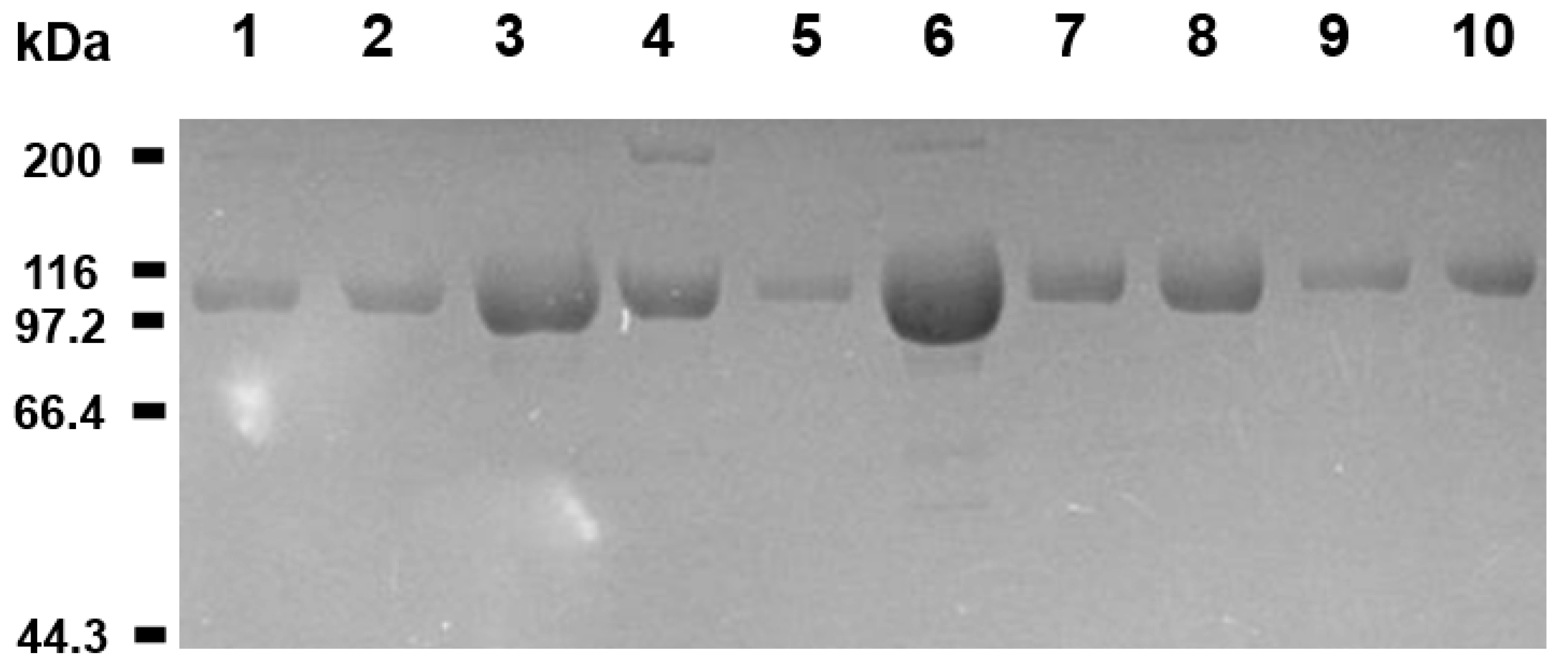
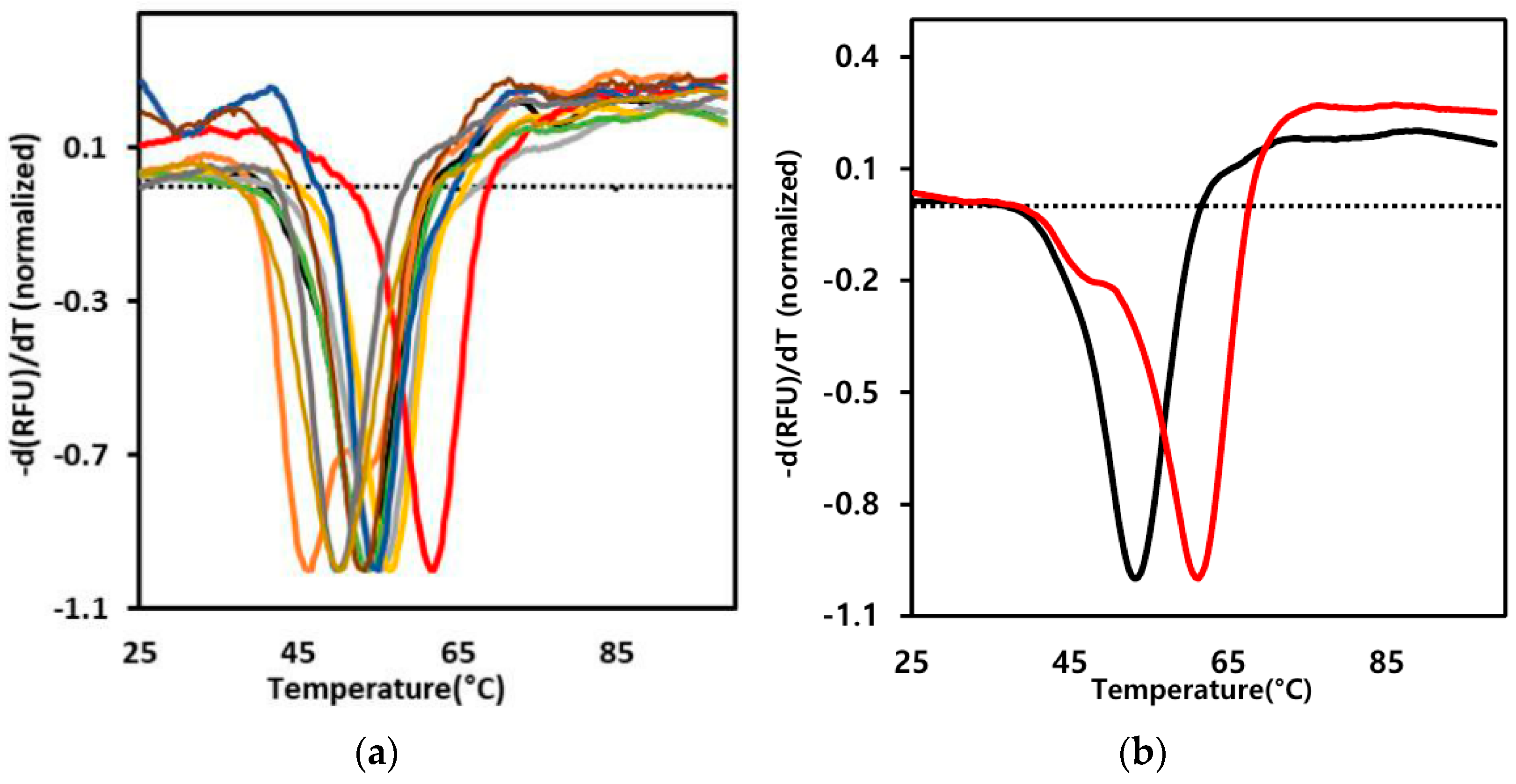
2.2. Screening for the Rmo-Stable Mutants
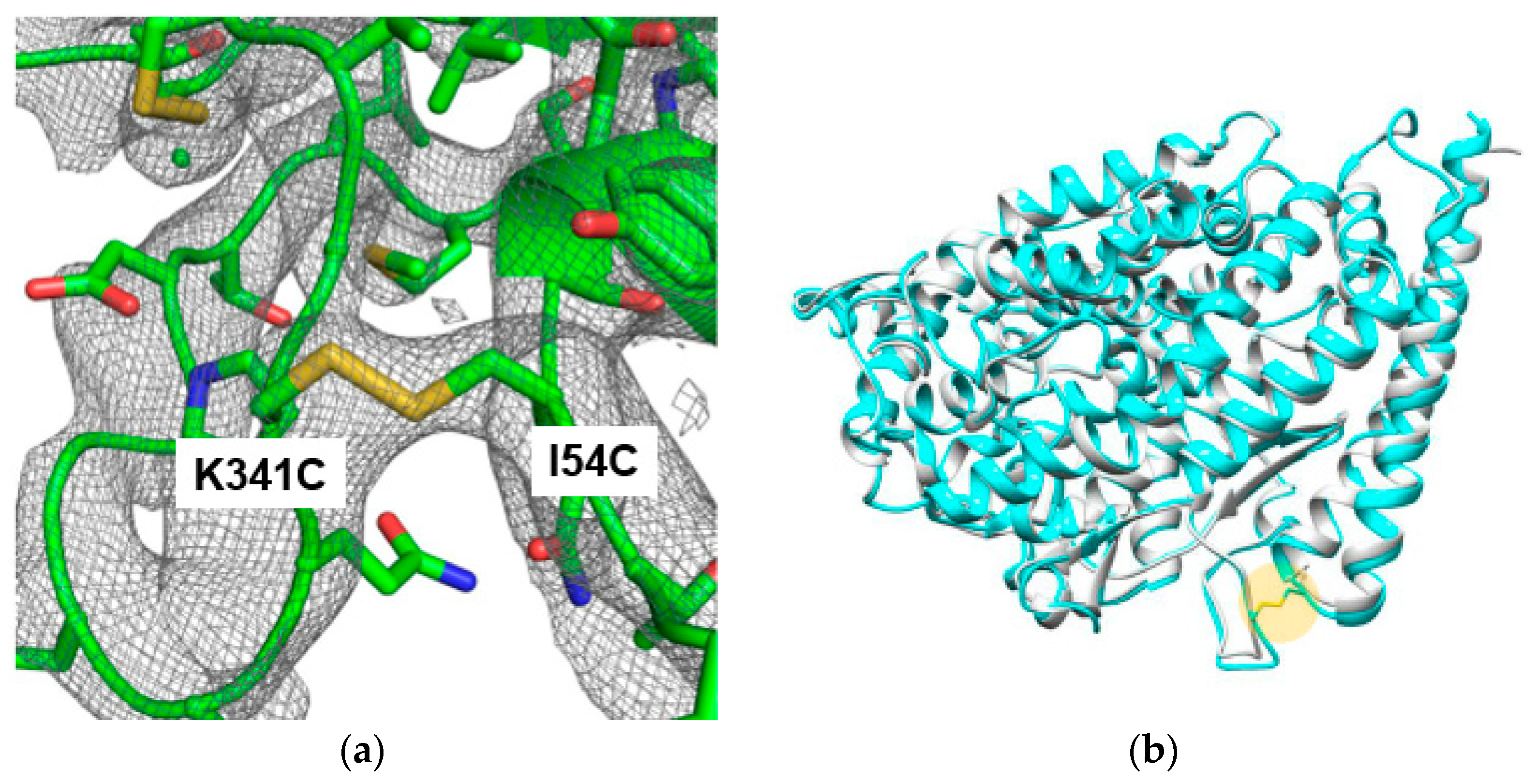
2.3. Structure Determination of the Thermostable Mutant
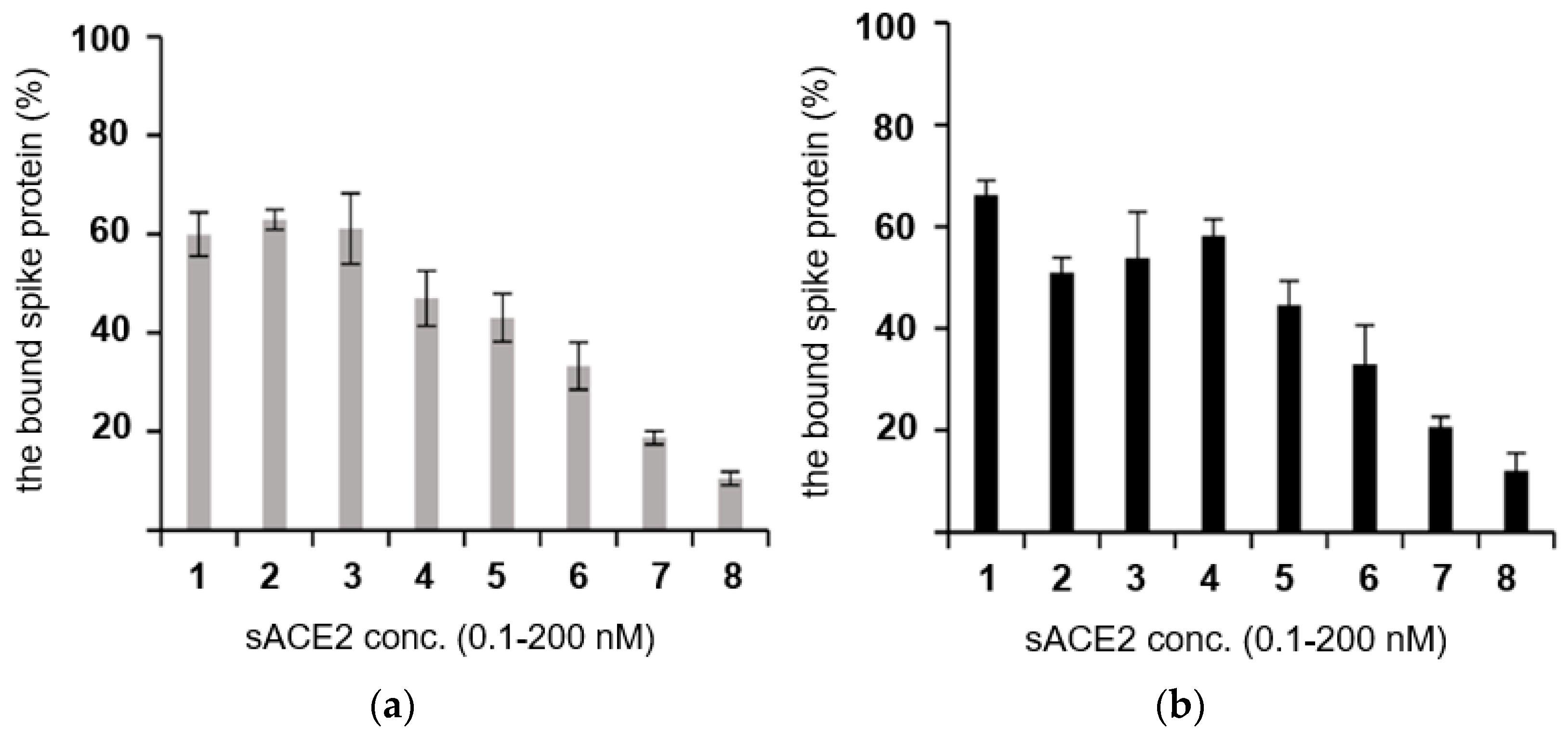
2.4. Binding to the Spike Protein
2.5. Cell-Based Competition Analysis
3. Materials and Methods
3.1. Cloning, Expression, and Purification of Proteins
3.2. Thermal Shift Assay
3.3. Crystallization and Structure Determination
3.4. Binding Affinity and Kinetic Analysis by SPR
3.5. Competitive Binding Assay by Cell-Based ELISA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Badawi, S.; Ali, B.R. ACE2 Nascence, trafficking, and SARS-CoV-2 pathogenesis: The saga continues. Hum. Genom. 2021, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Bank, S.; De, S.K.; Bankura, B.; Maiti, S.; Das, M.; Khan, G.A. ACE/ACE2 balance might be instrumental to explain the certain comorbidities leading to severe COVID-19 cases. Biosci. Rep. 2021, 41, BSR20202014. [Google Scholar] [CrossRef]
- Wysocki, J.; Ye, M.; Hassler, L.; Gupta, A.K.; Wang, Y.; Nicoleascu, V.; Randall, G.; Wertheim, J.A.; Batlle, D. A Novel Soluble ACE2 Variant with Prolonged Duration of Action Neutralizes SARS-CoV-2 Infection in Human Kidney Organoids. J. Am. Soc. Nephrol. 2021, 32, 795–803. [Google Scholar] [CrossRef]
- Iwanaga, N.; Cooper, L.; Rong, L.; Maness, N.J.; Beddingfield, B.; Qin, Z.; Crabtree, J.; Tripp, R.A.; Yang, H.; Blair, R.; et al. ACE2-IgG1 fusions with improved in vitro and in vivo activity against SARS-CoV-2. iScience 2022, 25, 103670. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Baker, S.C. Coronaviruses: From common colds to severe acute respiratory syndrome. Pediatr. Infect. Dis. J. 2004, 23, 1049–1050. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Iwanaga, N.; Cooper, L.; Rong, L.; Beddingfield, B.; Crabtree, J.; Tripp, R.A.; Qin, X.; Kolls, J.K. Novel ACE2-IgG1 fusions with improved in vitro and in vivo activity against SARS-CoV2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Renzi, F.; Seamann, A.; Ganguly, K.; Pandey, K.; Byrareddy, S.N.; Batra, S.; Kumar, S.; Ghersi, D. Engineering an ACE2-Derived Fragment as a Decoy for Novel SARS-CoV-2 Virus. ACS Pharmacol. Transl. Sci. 2023, 6, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Rudnik-Jansen, I.; Dinesen, A.; Selnihhin, D.; Mandrup, O.A.; Thiam, K.; Kjems, J.; Pedersen, F.S.; Howard, K.A. An albumin-angiotensin converting enzyme 2-based SARS-CoV-2 decoy with FcRn-driven half-life extension. Acta Biomater. 2022, 153, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Havranek, B.; Lindsey, G.W.; Higuchi, Y.; Itoh, Y.; Suzuki, T.; Okamoto, T.; Hoshino, A.; Procko, E.; Islam, S.M. A computationally designed ACE2 decoy has broad efficacy against SARS-CoV-2 omicron variants and related viruses in vitro and in vivo. Commun. Biol. 2023, 6, 513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Narayanan, K.K.; Cooper, L.; Chan, K.K.; Skeeters, S.S.; Devlin, C.A.; Aguhob, A.; Shirley, K.; Rong, L.; Rehman, J.; et al. An ACE2 decoy can be administered by inhalation and potently targets omicron variants of SARS-CoV-2. EMBO Mol. Med. 2022, 14, e16109. [Google Scholar] [CrossRef]
- Li, G.; Qian, K.; Zhang, S.; Fu, W.; Zhao, J.; Lei, C.; Hu, S. Engineered soluble ACE2 receptor: Responding to change with change. Front. Immunol. 2022, 13, 1084331. [Google Scholar] [CrossRef]
- Zekri, L.; Ruetalo, N.; Christie, M.; Walker, C.; Manz, T.; Rammensee, H.G.; Salih, H.R.; Schindler, M.; Jung, G. Novel ACE2 fusion protein with adapting activity against SARS-CoV-2 variants in vitro. Front. Immunol. 2023, 14, 1112505. [Google Scholar] [CrossRef] [PubMed]
- Kegler, A.; Drewitz, L.; Arndt, C.; Daglar, C.; Rodrigues Loureiro, L.; Mitwasi, N.; Neuber, C.; Gonzalez Soto, K.E.; Bartsch, T.; Baraban, L.; et al. A novel ACE2 decoy for both neutralization of SARS-CoV-2 variants and killing of infected cells. Front. Immunol. 2023, 14, 1204543. [Google Scholar] [CrossRef]
- Lu, M.; Yao, W.; Li, Y.; Ma, D.; Zhang, Z.; Wang, H.; Tang, X.; Wang, Y.; Li, C.; Cheng, D.; et al. Broadly Effective ACE2 Decoy Proteins Protect Mice from Lethal SARS-CoV-2 Infection. Microbiol. Spectr. 2023, 11, e0110023. [Google Scholar] [CrossRef]
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.A.; Rettko, N.J.; Zha, S.; Yamin, R.; et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 28046–28055. [Google Scholar] [CrossRef]
- Arimori, T.; Ikemura, N.; Okamoto, T.; Takagi, J.; Standley, D.M.; Hoshino, A. Engineering ACE2 decoy receptors to combat viral escapability. Trends Pharmacol. Sci. 2022, 43, 838–851. [Google Scholar] [CrossRef]
- Cianfarini, C.; Hassler, L.; Wysocki, J.; Hassan, A.; Nicolaescu, V.; Elli, D.; Gula, H.; Ibrahim, A.M.; Randall, G.; Henkin, J.; et al. Soluble Angiotensin-Converting Enzyme 2 Protein Improves Survival and Lowers Viral Titers in Lethal Mouse Model of Severe Acute Respiratory Syndrome Coronavirus Type 2 Infection with the Delta Variant. Cells 2024, 13, 203. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Consortium, C.-G.U.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.K.; Singh, R.B. SARS-CoV-2 variant biology and immune evasion. Prog. Mol. Biol. Transl. Sci. 2024, 202, 45–66. [Google Scholar] [CrossRef]
- Wysocki, J.; Ye, M.; Rodriguez, E.; Gonzalez-Pacheco, F.R.; Barrios, C.; Evora, K.; Schuster, M.; Loibner, H.; Brosnihan, K.B.; Ferrario, C.M.; et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: Prevention of angiotensin II-dependent hypertension. Hypertension 2010, 55, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Suzuki, T.; Arimori, T.; Ikemura, N.; Mihara, E.; Kirita, Y.; Ohgitani, E.; Mazda, O.; Motooka, D.; Nakamura, S.; et al. Engineered ACE2 receptor therapy overcomes mutational escape of SARS-CoV-2. Nat. Commun. 2021, 12, 3802. [Google Scholar] [CrossRef]
- Chan, M.C.; Chan, K.K.; Procko, E.; Shukla, D. Machine Learning Guided Design of High-Affinity ACE2 Decoys for SARS-CoV-2 Neutralization. J. Phys. Chem. B 2023, 127, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef]
- Schmidt, B.; Hogg, P.J. Search for allosteric disulfide bonds in NMR structures. BMC Struct. Biol. 2007, 7, 49. [Google Scholar] [CrossRef]
- Ferrari, M.; Mekkaoui, L.; Ilca, F.T.; Akbar, Z.; Bughda, R.; Lamb, K.; Ward, K.; Parekh, F.; Karattil, R.; Allen, C.; et al. Characterization of a Novel ACE2-Based Therapeutic with Enhanced Rather than Reduced Activity against SARS-CoV-2 Variants. J. Virol. 2021, 95, e0068521. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, M.; Chen, C.W.; Huang, J.K.; Zolkiewski, M.; Wen, L.; Krishnamoorthi, R. Disulfide bond effects on protein stability: Designed variants of Cucurbita maxima trypsin inhibitor-V. Protein Sci. 2001, 10, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Futami, J.; Miyamoto, A.; Hagimoto, A.; Suzuki, S.; Futami, M.; Tada, H. Evaluation of irreversible protein thermal inactivation caused by breakage of disulphide bonds using methanethiosulphonate. Sci. Rep. 2017, 7, 12471. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Functional aspects of protein flexibility. Cell Mol. Life Sci. 2009, 66, 2231–2247. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sACE2-Fc Mutants | Structural Rationale | Cα Distance (Å) | Tm (°C) | ΔTm (°C) |
|---|---|---|---|---|
| Wild type | na | na | 53.1 | 0 |
| N53C/Q340C (M1) | flexible loop stabilization | 5.9 | 56.6 | 3.5 |
| I54C/K341C (M2) | flexible loop stabilization | 6.0 | 61.2 | 8.1 |
| I21C/E87C (M3) | helix–loop stabilization | 6.6 | 45.8 | −7.3 |
| M62C/S47C (M4) | Helix–helix stabilization | 4.8 | 55.4 | 2.3 |
| A193C/V107C (M5) | helix kink–helix kink stabilization | 6.5 | 53.9 | 0.8 |
| V364C/V298C (M6) | helix–helix stabilization | 6.6 | 49.3 | −3.8 |
| S502C/R169C (M7) | helix–helix stabilization | 6.2 | 53.5 | 0.4 |
| N508C/S124C (M8) | helix–helix stabilization | 5.6 | 54.7 | 1.6 |
| A348C/H378C (M9) | helix–strand stabilization | 6.3 | 50.1 | −3.0 |
| Resolution range (Å) | 34.08–3.50 (3.63–3.50) * |
| Space group | H3 |
| Unit cell a, b, c (Å) α, β, γ (°) | 180.32, 180.32, 69.49 90.00, 90.00, 120.00 |
| Total reflections | 26,877 (2090) |
| Unique reflections | 9802 (912) |
| Redundancy | 2.7 (2.3) |
| Completeness (%) | 92.25 (86.5) |
| I/σI | 8.67 (4.52) |
| Rmerge (%) | 9.7 (20.5) |
| CC1/2 | 0.984 (0.885) |
| Reflections used in refinement | 9799 (912) |
| Reflections used for R-free | 983 (91) |
| R-work/R-free | 20.0 (23.2)/25.9 (29.5) |
| Number of total atoms | 4866 |
| Protein residues | 597 |
| RMS (bonds) (Å) | 0.031 |
| RMS (angles) (°) | 0.8 |
| Ramachandran plot (%) Favored/allowed/outliers | 93.95/5.88/0.17 |
| Average B-factor (Å2) | 62.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.S.; Kim, M.; Park, H.M.; Kim, H.J.; Ryu, S.E. Disulfide Bond Engineering of Soluble ACE2 for Thermal Stability Enhancement. Int. J. Mol. Sci. 2024, 25, 9919. https://doi.org/10.3390/ijms25189919
Kim YS, Kim M, Park HM, Kim HJ, Ryu SE. Disulfide Bond Engineering of Soluble ACE2 for Thermal Stability Enhancement. International Journal of Molecular Sciences. 2024; 25(18):9919. https://doi.org/10.3390/ijms25189919
Chicago/Turabian StyleKim, Yoon Soo, Myeongbin Kim, Hye Min Park, Hyun Jin Kim, and Seong Eon Ryu. 2024. "Disulfide Bond Engineering of Soluble ACE2 for Thermal Stability Enhancement" International Journal of Molecular Sciences 25, no. 18: 9919. https://doi.org/10.3390/ijms25189919