
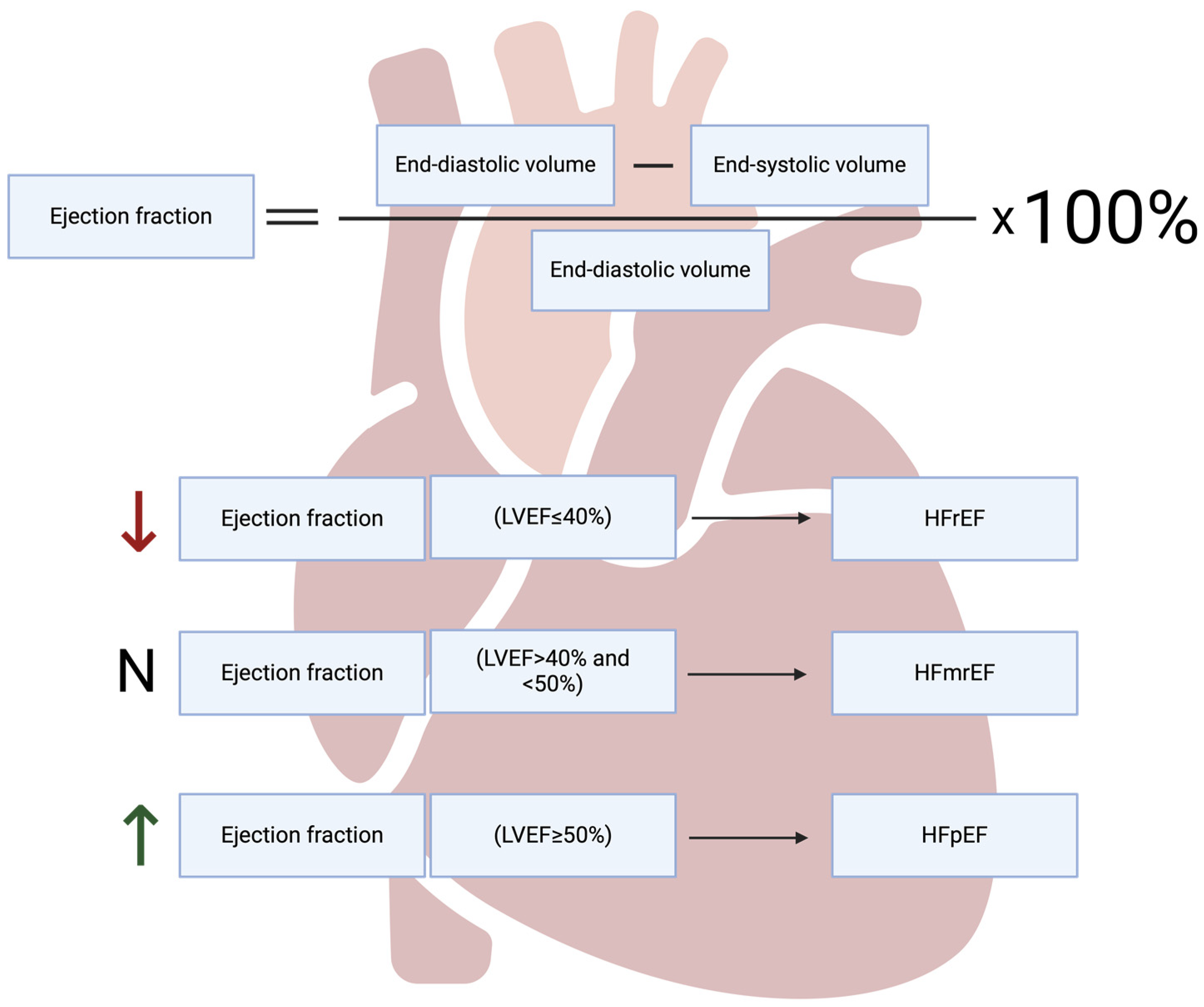
The Role of Programmed Types of Cell Death in Pathogenesis of Heart Failure with Preserved Ejection Fraction

 , , ,
, , ,
Abstract
:1. Introduction
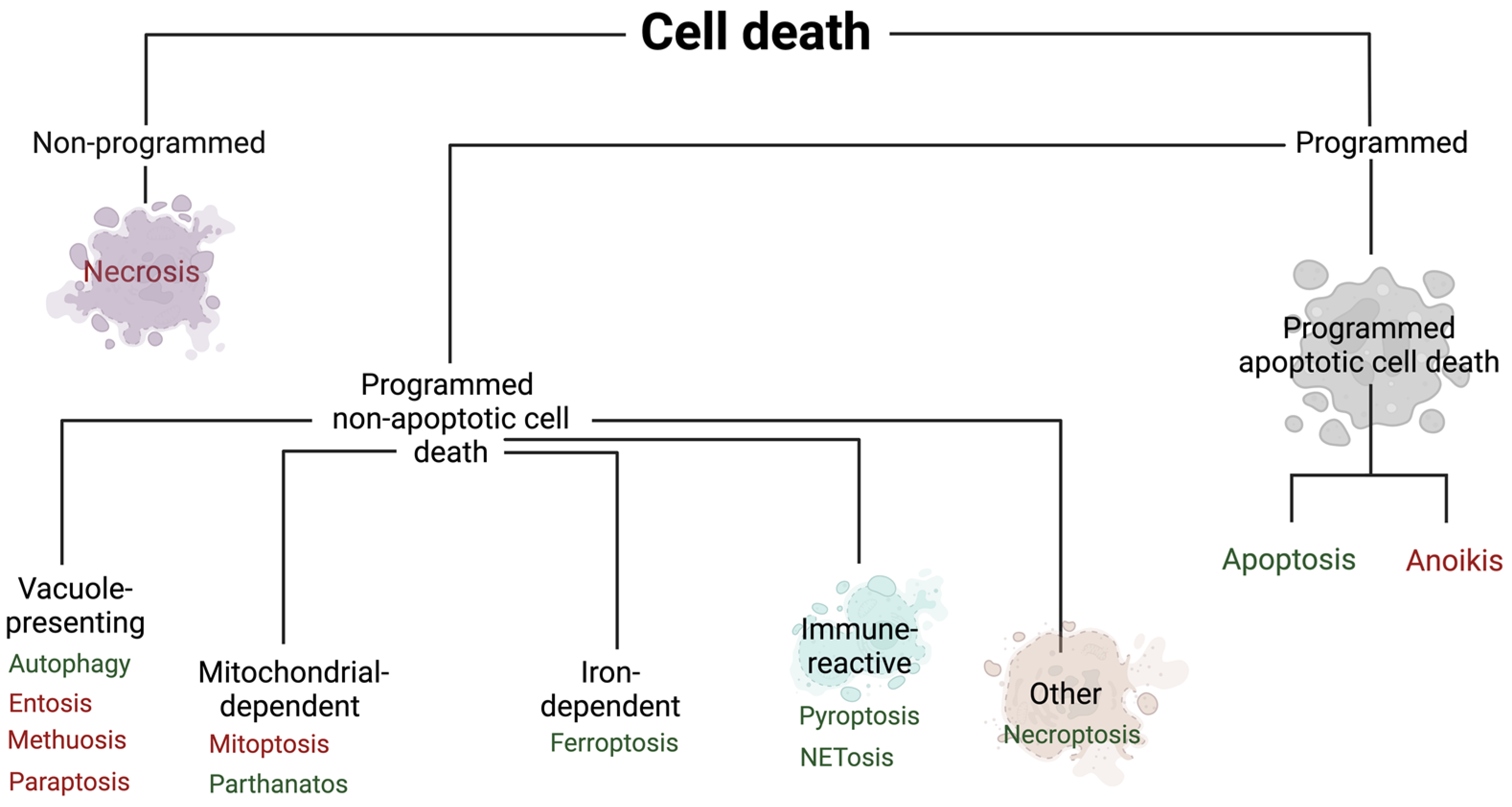
2. Non-Programmed Cell Death
3. Programmed Cell Death
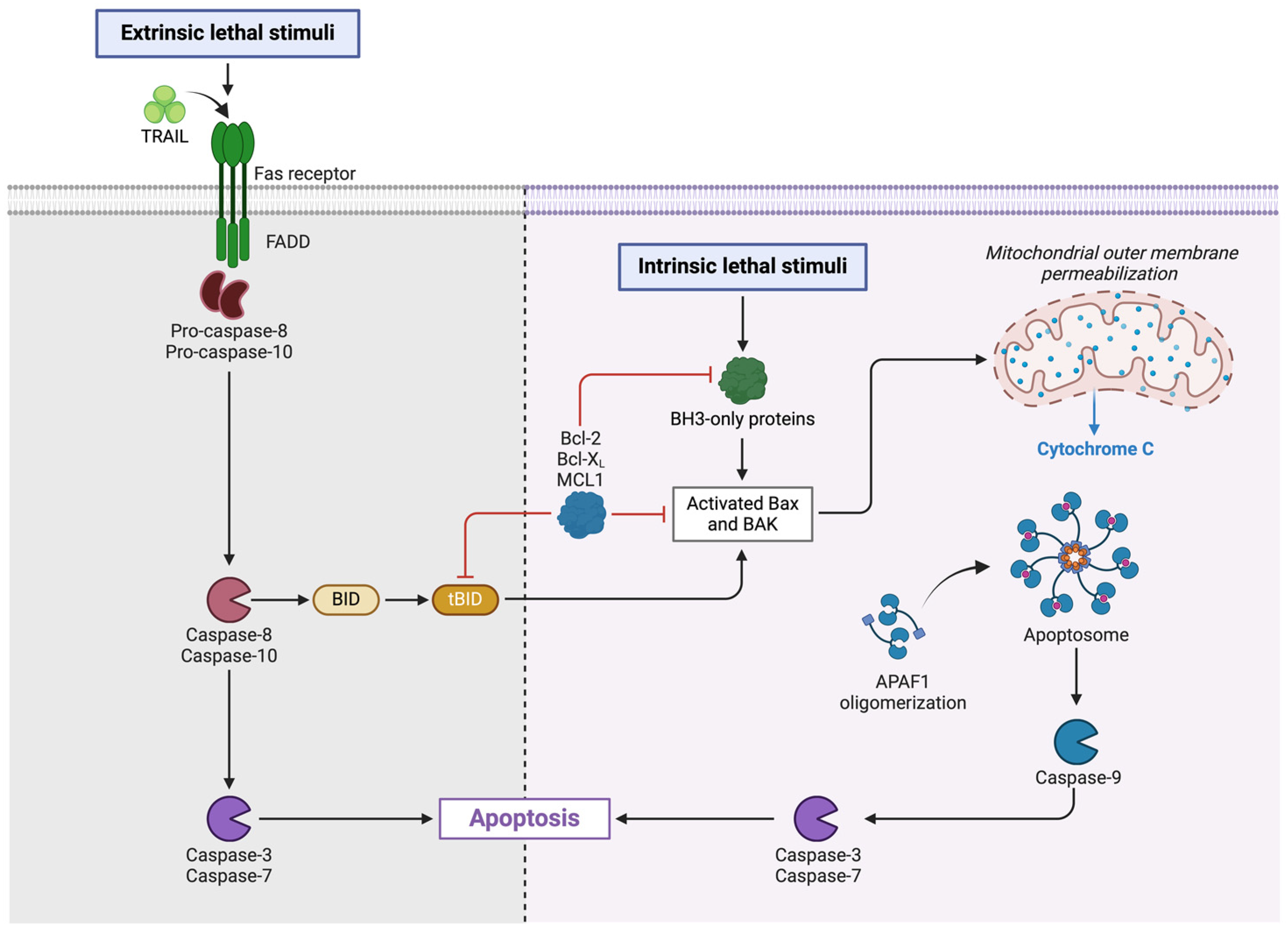
3.1. Programmed Apoptotic Cell Death
3.1.1. Apoptosis
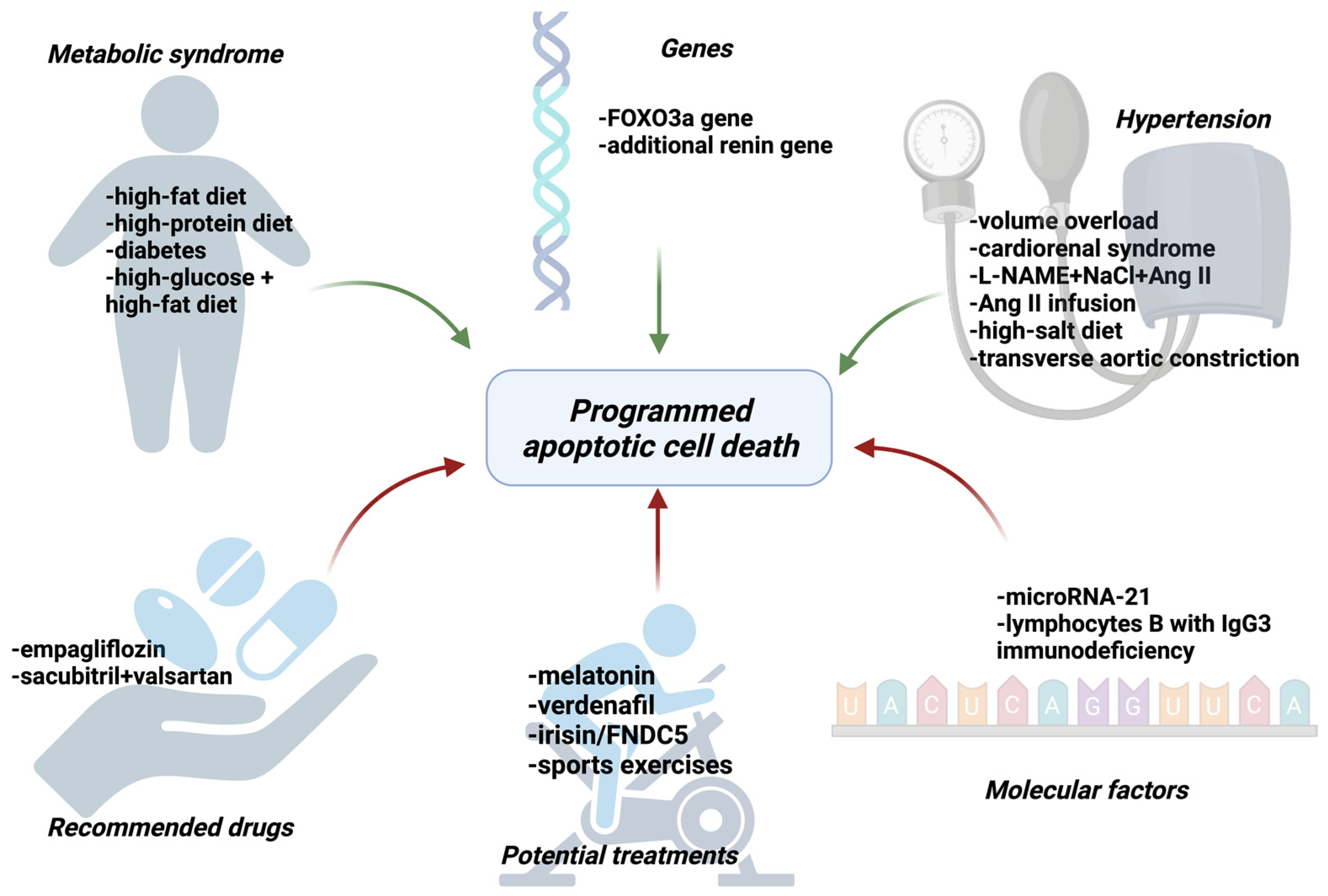
Apoptosis Plays a Role in HF and HFpEF
Drugs Recommended in HFpEF Reduce Apoptosis
Searching for New Treatment Options with a Potential Target in Apoptosis
Other Targets of Drugs (Genes or Proteins)
Metabolic Syndrome Enhances Apoptosis
Hypertension Enhances Apoptosis
3.2. Programmed Non-Apoptotic Cell Death
3.2.1. Ferroptosis
3.2.2. Autophagy
3.2.3. Parathantos
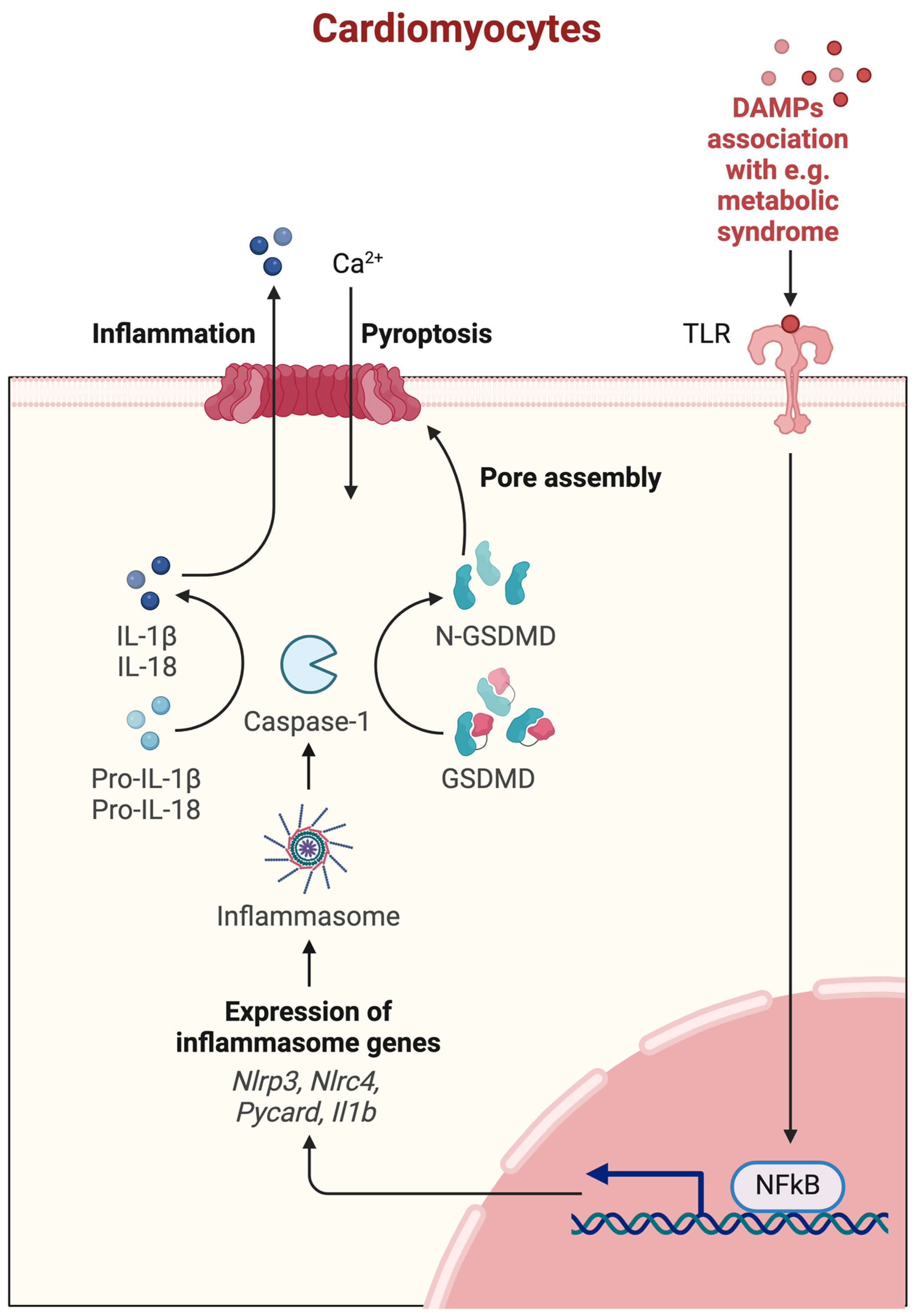
3.2.4. Pyroptosis
3.2.5. NETosis
NETs Induce Aseptic Inflammatory Process Associated with HFpEF
Seipin Knockout Leads to NET Formation, Interstitial Fibrosis, and Ventricular Stiffness
Further Attempts: Modifying the Potent Signal Paths of NETosis in HF Mouse Models
3.2.6. Necroptosis
The Role of RIPK3-MLKL Pathway in HFpEF
Caspase-8 Is Responsible for the Balance between Apoptosis and Necroptosis
Necroptosis Plays an Important Role in HFpEF
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Borlaug, B.A.; Paulus, W.J. Heart failure with preserved ejection fraction: Pathophysiology, diagnosis, and treatment. Eur. Heart J. 2011, 32, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features. World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arter. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Pagel, I.; Höhnel, K.; Lutz, J.; Buttgereit, J.; Langenickel, T.; Hamet, P.; Dietz, R.; Willenbrock, R. Regulation of caspase 3 and Fas in pressure overload-induced left ventricular dysfunction. Eur. J. Heart Fail. 2004, 6, 845–851. [Google Scholar] [CrossRef]
- Kitahori, K.; He, H.; Kawata, M.; Cowan, D.B.; Friehs, I.; del Nido, P.J.; McGowan, F.X. Development of left ventricular diastolic dysfunction with preservation of ejection fraction during progression of infant right ventricular hypertrophy. Circ. Heart Fail. 2009, 2, 599–607. [Google Scholar] [CrossRef]
- Oatmen, K.E.; Cull, E.; Spinale, F.G. Heart failure as interstitial cancer: Emergence of a malignant fibroblast phenotype. Nat. Rev. Cardiol. 2020, 17, 523–531. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, H.; Gao, J.; Liu, Y.; Li, J.; Wang, J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell. Cardiol. 2019, 136, 27–41. [Google Scholar] [CrossRef]
- Moorjani, N.; Westaby, S.; Narula, J.; Catarino, P.A.; Brittin, R.; Kemp, T.J.; Narula, N.; Sugden, P.H. Effects of left ventricular volume overload on mitochondrial and death-receptor–mediated apoptotic pathways in the transition to heart failure. Am. J. Cardiol. 2009, 103, 1261–1268. [Google Scholar] [CrossRef]
- Kresoja, K.; Rommel, K.; Wachter, R.; Henger, S.; Besler, C.; Klöting, N.; Schnelle, M.; Hoffmann, A.; Büttner, P.; Ceglarek, U.; et al. Proteomics to improve phenotyping in obese patients with heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2021, 23, 1633–1644. [Google Scholar] [CrossRef]
- Hage, C.; Michaëlsson, E.; Linde, C.; Donal, E.; Daubert, J.C.; Gan, L.M.; Lund, L.H. Inflammatory Biomarkers Predict Heart Failure Severity and Prognosis in Patients With Heart Failure With Preserved Ejection Fraction: A Holistic Proteomic Approach. Circ. Cardiovasc. Genet. 2017, 10, e001633. [Google Scholar] [CrossRef]
- Yang, C.C.; Chen, Y.T.; Wallace, C.G.; Chen, K.H.; Cheng, B.C.; Sung, P.H.; Li, Y.C.; Ko, S.F.; Chang, H.W.; Yip, H.K. Early administration of empagliflozin preserved heart function in cardiorenal syndrome in rat. Biomed. Pharmacother. 2019, 109, 658–670. [Google Scholar] [CrossRef]
- Yeh, J.-N.; Yue, Y.; Chu, Y.-C.; Huang, C.-R.; Yang, C.-C.; Chiang, J.Y.; Yip, H.-K.; Guo, J. Entresto protected the cardiomyocytes and preserved heart function in cardiorenal syndrome rat fed with high-protein diet through regulating the oxidative stress and Mfn2-mediated mitochondrial functional integrity. Biomed. Pharmacother. 2021, 144, 112244. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, L.N.; Guo, S.; Zhao, X.Y.; Liu, Y.Z.; Liang, C.; Tu, S.; Wang, D.; Li, L.; Dong, J.Z.; et al. Melatonin improves cardiac function in a mouse model of heart failure with preserved ejection fraction. Redox Biol. 2018, 18, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Mátyás, C.; Németh, B.T.; Oláh, A.; Török, M.; Ruppert, M.; Kellermayer, D.; Barta, B.A.; Szabó, G.; Kökény, G.; Horváth, E.M.; et al. Prevention of the development of heart failure with preserved ejection fraction by the phosphodiesterase-5A inhibitor vardenafil in rats with type 2 diabetes. Eur. J. Heart Fail. 2017, 19, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Guo, Y.; Xia, Y.; Li, C.; Xu, X.; Qi, T.; Zhang, F.; Fan, M.; Hu, G.; Zhao, H.; et al. FNDC5/Irisin attenuates diabetic cardiomyopathy in a type 2 diabetes mouse model by activation of integrin alphaV/beta5-AKT signaling and reduction of oxidative/nitrosative stress. J. Mol. Cell. Cardiol. 2021, 160, 27–41. [Google Scholar] [CrossRef]
- Wang, H.; Bei, Y.; Lu, Y.; Sun, W.; Liu, Q.; Wang, Y.; Cao, Y.; Chen, P.; Xiao, J.; Kong, X. Exercise Prevents Cardiac Injury and Improves Mitochondrial Biogenesis in Advanced Diabetic Cardiomyopathy with PGC-1α and Akt Activation. Cell. Physiol. Biochem. 2015, 35, 2159–2168. [Google Scholar] [CrossRef]
- Dai, B.; Li, H.; Fan, J.; Zhao, Y.; Yin, Z.; Nie, X.; Wang, D.W.; Chen, C. MiR-21 protected against diabetic cardiomyopathy induced diastolic dysfunction by targeting gelsolin. Cardiovasc. Diabetol. 2018, 17, 123. [Google Scholar] [CrossRef]
- Dong, S.; Ma, W.; Hao, B.; Hu, F.; Yan, L.; Yan, X.; Wang, Y.; Chen, Z.; Wang, Z. microRNA-21 promotes cardiac fibrosis and development of heart failure with preserved left ventricular ejection fraction by up-regulating Bcl-2. Int. J. Clin. Exp. Pathol. 2014, 7, 565–574. [Google Scholar]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease with Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Connelly, K.; Kelly, D.; Zhang, Y.; Prior, D.; Martin, J.; Cox, A.; Thai, K.; Feneley, M.; Tsoporis, J.; White, K.; et al. Functional, structural and molecular aspects of diastolic heart failure in the diabetic (mRen-2)27 rat. Cardiovasc. Res. 2007, 76, 280–291. [Google Scholar] [CrossRef]
- Cordero-Reyes, A.M.; Youker, K.A.; Trevino, A.R.; Celis, R.; Hamilton, D.J.; Flores-Arredondo, J.H.; Orrego, C.M.; Bhimaraj, A.; Estep, J.D.; Torre-Amione, G. Full Expression of Cardiomyopathy Is Partly Dependent on B-Cells: A Pathway That Involves Cytokine Activation, Immunoglobulin Deposition, and Activation of Apoptosis. J. Am. Heart Assoc. 2016, 5, e002484. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Kajstura, J.; Chimenti, C.; Jakoniuk, I.; Leri, A.; Maseri, A.; Nadal-Ginard, B.; Anversa, P. Myocardial cell death in human diabetes. Circ. Res. 2000, 87, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Chen, J.; Wu, C.; Jiang, P.; Wang, Y.; Zhang, Y.; Jiang, Y.; Li, X. Deciphering the Protein, Modular Connections and Precision Medicine for Heart Failure with Preserved Ejection Fraction and Hypertension Based on TMT Quantitative Proteomics and Molecular Docking. Front. Physiol. 2021, 12, 607089. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Flamini, S.; Cozzolino, A.; Bereshchenko, O.; Ronchetti, S.; Cianflone, E.; Gagliardi, A.; Ricci, E.; Rafaniello, C.; et al. Deficit of glucocorticoid-induced leucine zipper amplifies angiotensin-induced cardiomyocyte hypertrophy and diastolic dysfunction. J. Cell. Mol. Med. 2021, 25, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.M.; Nguyen, I.T.N.; Krebber, M.M.; van de Wouw, J.; Mokry, M.; Cramer, M.J.; Duncker, D.J.; Verhaar, M.C.; Joles, J.A.; Cheng, C. Limited synergy of obesity and hypertension, prevalent risk factors in onset and progression of heart failure with preserved ejection fraction. J. Cell. Mol. Med. 2019, 23, 6666–6678. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, H.; Yao, W.; Li, L.; Niu, P.; Huo, Y.; Tan, W. Morphometric, Hemodynamic, and Multi-Omics Analyses in Heart Failure Rats with Preserved Ejection Fraction. Int. J. Mol. Sci. 2020, 21, 3362. [Google Scholar] [CrossRef]
- Niessner, A.; Hohensinner, P.J.; Rychli, K.; Neuhold, S.; Zorn, G.; Richter, B.; Hülsmann, M.; Berger, R.; Mörtl, D.; Huber, K.; et al. Prognostic value of apoptosis markers in advanced heart failure patients. Eur. Heart J. 2009, 30, 789–796. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef]
- Yu, Q.; Kou, W.; Zhou, S.; Luan, P.; Xu, X.; Li, H.; Zhuang, J.; Wang, J.; Zhao, Y.; Xu, Y.; et al. FNDC5/Irisin inhibits pathological cardiac hypertrophy. Clin. Sci. 2019, 133, 611–627. [Google Scholar] [CrossRef]
- Kwak, H.B.; Song, W.; Lawler, J.M. Exercise training attenuates age-induced elevation in Bax/Bcl-2 ratio, apoptosis, and remodeling in the rat heart. FASEB J. 2006, 20, 791–793. [Google Scholar] [CrossRef]
- Tao, L.; Huang, X.; Xu, M.; Qin, Z.; Zhang, F.; Hua, F.; Jiang, X.; Wang, Y. Value of circulating miRNA-21 in the diagnosis of subclinical diabetic cardiomyopathy. Mol. Cell. Endocrinol. 2020, 518, 110944. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.A.; Mauro, A.G.; Carbone, S.; Marchetti, C.; Gill, R.; Mezzaroma, E.; Raleigh, J.V.; Salloum, F.N.; Van Tassell, B.W.; Abbate, A.; et al. A mouse model of heart failure with preserved ejection fraction due to chronic infusion of a low subpressor dose of angiotensin II. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H771–H778. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; He, L.-L.; Zhang, G.-R.; Zuo, Q.-J.; Wang, Z.-L.; Zhai, J.-L.; Zhang, T.-T.; Wang, Y.; Ma, H.-J.; Guo, Y.-F. Canagliflozin mitigates ferroptosis and ameliorates heart failure in rats with preserved ejection fraction. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2022, 395, 945–962. [Google Scholar] [CrossRef]
- Kitakata, H.; Endo, J.; Hashimoto, S.; Mizuno, E.; Moriyama, H.; Shirakawa, K.; Goto, S.; Katsumata, Y.; Fukuda, K.; Sano, M. Imeglimin prevents heart failure with preserved ejection fraction by recovering the impaired unfolded protein response in mice subjected to cardiometabolic stress. Biochem. Biophys. Res. Commun. 2021, 572, 185–190. [Google Scholar] [CrossRef]
- Zhang, L.-L.; Chen, G.-H.; Tang, R.-J.; Xiong, Y.-Y.; Pan, Q.; Jiang, W.-Y.; Gong, Z.-T.; Chen, C.; Li, X.-S.; Yang, Y.-J. Levosimendan Reverses Cardiac Malfunction and Cardiomyocyte Ferroptosis During Heart Failure with Preserved Ejection Fraction via Connexin 43 Signaling Activation. Cardiovasc. Drugs Ther. 2023, 38, 705–718. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Song, J.; Xie, M.; Liu, Y.; Dong, Z.; Liu, X.; Li, X.; Zhang, M.; Chen, Y.; et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic. Biol. Med. 2022, 181, 130–142. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Zhou, W.; Men, H.; Bao, T.; Sun, Y.; Wang, Q.; Tan, Y.; Keller, B.B.; Tong, Q.; et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm. Sin. B 2022, 12, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Cantrell, A.C.; Chen, J.-X.; Gu, W.; Zeng, H. SIRT3 Deficiency Enhances Ferroptosis and Promotes Cardiac Fibrosis via p53 Acetylation. Cells 2023, 12, 1428. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Adarsh, K.; Kaur, H.; Mohan, V. Coenzyme Q10 (CoQ10) in isolated diastolic heart failure in hypertrophic cardiomyopathy (HCM). Biofactors 2008, 32, 145–149. [Google Scholar] [CrossRef]
- Pierce, J.D.; Shen, Q.; Mahoney, D.E.; Rahman, F.; Krueger, K.J.; Diaz, F.J.; Clark, L.; Smith, C.; Vacek, J.; Hiebert, J.B. Effects of Ubiquinol and/or D-ribose in Patients With Heart Failure With Preserved Ejection Fraction. Am. J. Cardiol. 2022, 176, 79–88. [Google Scholar] [CrossRef]
- Sobirin, M.A.; Herry, Y.; Sofia, S.N.; Uddin, I.; Rifqi, S.; Tsutsui, H. Effects of coenzyme Q10 supplementation on diastolic function in patients with heart failure with preserved ejection fraction. Drug Discov. Ther. 2019, 13, 38–46. [Google Scholar] [CrossRef]
- Samuel, T.Y.; Hasin, T.; Gotsman, I.; Weitzman, T.; Ben Ivgi, F.; Dadon, Z.; Asher, E.; Amir, O.; Glikson, M.; Alcalai, R.; et al. Coenzyme Q10 in the Treatment of Heart Failure with Preserved Ejection Fraction: A Prospective, Randomized, Double-Blind, Placebo-Controlled Trial. Drugs R&D 2022, 22, 25–33. [Google Scholar] [CrossRef]
- Xu, J.; Kitada, M.; Ogura, Y.; Liu, H.; Koya, D. Dapagliflozin Restores Impaired Autophagy and Suppresses Inflammation in High Glucose-Treated HK-2 Cells. Cells 2021, 10, 1457. [Google Scholar] [CrossRef]
- Xie, Z.; Lau, K.; Eby, B.; Lozano, P.; He, C.; Pennington, B.; Li, H.; Rathi, S.; Dong, Y.; Tian, R.; et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011, 60, 1770–1778. [Google Scholar] [CrossRef]
- Wu, S.; Yu, W.; Jiang, X.; Huang, R.; Zhang, X.; Lan, J.; Zhong, G.; Wan, F.; Tang, Z.; Hu, L. Protective effects of curcumin on ATO-induced nephrotoxicity in ducks in relation to suppressed autophagy, apoptosis and dyslipidemia by regulating oxidative stress. Ecotoxicol. Environ. Saf. 2021, 219, 112350. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, S.; Zhou, S.; Yan, X.; Wang, Y.; Chen, J.; Mellen, N.; Kong, M.; Gu, J.; Tan, Y.; et al. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. J. Mol. Cell. Cardiol. 2014, 77, 42–52. [Google Scholar] [CrossRef]
- Wang, Q.; Ren, J. mTOR-Independent autophagy inducer trehalose rescues against insulin resistance-induced myocardial contractile anomalies: Role of p38 MAPK and Foxo1. Pharmacol. Res. 2016, 111, 357–373. [Google Scholar] [CrossRef]
- Liaudet, L.; Szabó, E.; Timashpolsky, L.; Virág, L.; Cziráki, A.; Szabó, C. Suppression of poly (ADP-ribose) polymerase activation by 3-aminobenzamide in a rat model of myocardial infarction: Long-term morphological and functional consequences. Br. J. Pharmacol. 2001, 133, 1424–1430. [Google Scholar] [CrossRef]
- Grupp, I.L.; Jackson, T.M.; Hake, P.; Grupp, G.; Szabó, C. Protection against hypoxia-reoxygenation in the absence of poly (ADP-ribose) synthetase in isolated working hearts. J. Mol. Cell. Cardiol. 1999, 31, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-Z.; Swanson, R.A.; Simonis, U.; Ma, X.; Cecchini, G.; Gray, M.O. Poly(ADP-ribose) polymerase-1 hyperactivation and impairment of mitochondrial respiratory chain complex I function in reperfused mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H714–H723. [Google Scholar] [CrossRef]
- Docherty, J.C.; Kuzio, B.; A Silvester, J.; Bowes, J.; Thiemermann, C. An inhibitor of poly (ADP-ribose) synthetase activity reduces contractile dysfunction and preserves high energy phosphate levels during reperfusion of the ischaemic rat heart. Br. J. Pharmacol. 1999, 127, 1518–1524. [Google Scholar] [CrossRef]
- Pillai, J.B.; Russell, H.M.; Raman, J.; Jeevanandam, V.; Gupta, M.P. Increased expression of poly(ADP-ribose) polymerase-1 contributes to caspase-independent myocyte cell death during heart failure. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H486–H496. [Google Scholar] [CrossRef]
- Xiao, C.-Y.; Chen, M.; Zsengellér, Z.; Li, H.; Kiss, L.; Kollai, M.; Szabó, C. Poly(ADP-Ribose) polymerase promotes cardiac remodeling, contractile failure, and translocation of apoptosis-inducing factor in a murine experimental model of aortic banding and heart failure. J. Pharmacol. Exp. Ther. 2005, 312, 891–898. [Google Scholar] [CrossRef]
- Barany, T.; Simon, A.; Szabo, G.; Benko, R.; Mezei, Z.; Molnar, L.; Becker, D.; Merkely, B.; Zima, E.; Horvath, E.M. Oxidative Stress-Related Parthanatos of Circulating Mononuclear Leukocytes in Heart Failure. Oxidative Med. Cell. Longev. 2017, 2017, 1249614. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.-Y.; Shi, Y.; Li, Z.; Li, H.; Wu, L.-D.; Zhou, W.-Y.; Gu, Y.; Ling, Z.-Y.; Zhang, J.-X.; Chen, S.-L. Involvement of pyroptosis pathway in epicardial adipose tissue—Myocardium axis in experimental heart failure with preserved ejection fraction. Biochem. Biophys. Res. Commun. 2022, 636 Pt 2, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.E.; Tromp, J.; Shah, S.J.; Lam, C.S.P.; Lewis, G.D.; A Borlaug, B.; Sharma, K.; Pandey, A.; Sweitzer, N.K.; Kitzman, D.W.; et al. Phenomapping in heart failure with preserved ejection fraction: Insights, limitations, and future directions. Cardiovasc. Res. 2023, 118, 3403–3415. [Google Scholar] [CrossRef] [PubMed]
- Koepp, K.E.; Obokata, M.; Reddy, Y.N.; Olson, T.P.; Borlaug, B.A. Hemodynamic and Functional Impact of Epicardial Adipose Tissue in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2020, 8, 657–666. [Google Scholar] [CrossRef]
- Mahabadi, A.A.; Anapliotis, V.; Dykun, I.; Hendricks, S.; Al-Rashid, F.; Lüdike, P.; Totzeck, M.; Rassaf, T. Epicardial fat and incident heart failure with preserved ejection fraction in patients with coronary artery disease. Int. J. Cardiol. 2022, 357, 140–145. [Google Scholar] [CrossRef]
- Ling, S.; Xu, J.-W. NETosis as a Pathogenic Factor for Heart Failure. Oxidative Med. Cell. Longev. 2021, 2021, 6687096. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Hage, C.; Michaëlsson, E.; Kull, B.; Miliotis, T.; Svedlund, S.; Linde, C.; Donal, E.; Daubert, J.; Gan, L.; Lund, L.H. Myeloperoxidase and related biomarkers are suggestive footprints of endothelial microvascular inflammation in HFpEF patients. ESC Heart Fail. 2020, 7, 1534–1546. [Google Scholar] [CrossRef] [PubMed]
- Boralkar, K.A.; Kobayashi, Y.; Amsallem, M.; Ataam, J.A.; Moneghetti, K.J.; Cauwenberghs, N.; Horne, B.D.; Knowlton, K.U.; Maecker, H.; Kuznetsova, T.; et al. Value of Neutrophil to Lymphocyte Ratio and Its Trajectory in Patients Hospitalized with Acute Heart Failure and Preserved Ejection Fraction. Am. J. Cardiol. 2020, 125, 229–235. [Google Scholar] [CrossRef]
- Pagac, M.; Cooper, D.E.; Qi, Y.; Lukmantara, I.E.; Mak, H.Y.; Wu, Z.; Tian, Y.; Liu, Z.; Lei, M.; Du, X.; et al. SEIPIN Regulates Lipid Droplet Expansion and Adipocyte Development by Modulating the Activity of Glycerol-3-phosphate Acyltransferase. Cell Rep. 2016, 17, 1546–1559. [Google Scholar] [CrossRef]
- Bai, B.; Yang, W.; Fu, Y.; Foon, H.L.; Tay, W.T.; Yang, K.; Luo, C.; Gunaratne, J.; Lee, P.; Zile, M.R.; et al. Seipin Knockout Mice Develop Heart Failure With Preserved Ejection Fraction. JACC Basic Transl. Sci. 2019, 4, 924–937. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Gola, A.; Winkelmann, M.; Jakob, S.M.; Groesser, L.; Borgolte, J.; Pogoda, F.; Pick, R.; Pruenster, M.; Müller-Höcker, J.; et al. The cytokine midkine supports neutrophil trafficking during acute inflammation by promoting adhesion via beta2 integrins (CD11/CD18). Blood 2014, 123, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, L.T.; Grabmaier, U.; Uhl, A.; Gess, S.; Boehm, F.; Zehrer, A.; Pick, R.; Salvermoser, M.; Czermak, T.; Pircher, J.; et al. Midkine drives cardiac inflammation by promoting neutrophil trafficking and NETosis in myocarditis. J. Exp. Med. 2019, 216, 350–368. [Google Scholar] [CrossRef]
- Martinod, K.; Witsch, T.; Erpenbeck, L.; Savchenko, A.; Hayashi, H.; Cherpokova, D.; Gallant, M.; Mauler, M.; Cifuni, S.M.; Wagner, D.D. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J. Exp. Med. 2017, 214, 439–458. [Google Scholar] [CrossRef]
- Pedersen, F.; Waschki, B.; Marwitz, S.; Goldmann, T.; Kirsten, A.; Malmgren, A.; Rabe, K.F.; Uddin, M.; Watz, H. Neutrophil extracellular trap formation is regulated by CXCR2 in COPD neutrophils. Eur. Respir. J. 2018, 51, 1700970. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.-L.; Lin, Q.-Y.; Liu, Y.; Guan, X.-M.; Ma, X.-L.; Cao, H.-J.; Liu, Y.; Bai, J.; Xia, Y.-L.; et al. CXCL1–CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur. Heart J. 2018, 39, 1818–1831. [Google Scholar] [CrossRef]
- Desai, J.; Kumar, S.V.; Mulay, S.R.; Konrad, L.; Romoli, S.; Schauer, C.; Herrmann, M.; Bilyy, R.; Müller, S.; Popper, B.; et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur. J. Immunol. 2016, 46, 223–229. [Google Scholar] [CrossRef]
- Amini, P.; Stojkov, D.; Wang, X.; Wicki, S.; Kaufmann, T.; Wong, W.W.; Simon, H.; Yousefi, S. NET formation can occur independently of RIPK3 and MLKL signaling. Eur. J. Immunol. 2016, 46, 178–184. [Google Scholar] [CrossRef]
- Zhu, P.; Wan, K.; Yin, M.; Hu, P.; Que, Y.; Zhou, X.; Zhang, L.; Li, T.; Du, Y.; Xu, G.; et al. RIPK3 Induces Cardiomyocyte Necroptosis via Inhibition of AMPK-Parkin-Mitophagy in Cardiac Remodelling after Myocardial Infarction. Oxidative Med. Cell. Longev. 2021, 2021, 6635955. [Google Scholar] [CrossRef] [PubMed]
- Zhe-Wei, S.; Li-Sha, G.; Yue-Chun, L. The Role of Necroptosis in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 721. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhang, J.; Qian, J.; Wang, X.; Zhang, W.; Chen, X. Ca2+/Calmodulin-Dependent Protein Kinase II Regulation by RIPK3 Alleviates Necroptosis in Transverse Arch Constriction-Induced Heart Failure. Front. Cardiovasc. Med. 2022, 9, 847362. [Google Scholar] [CrossRef]
- Zi, M.; Stafford, N.; Prehar, S.; Baudoin, F.; Oceandy, D.; Wang, X.; Bui, T.; Shaheen, M.; Neyses, L.; Cartwright, E.J. Cardiac hypertrophy or failure?—A systematic evaluation of the transverse aortic constriction model in C57BL/6NTac and C57BL/6J substrains. Curr. Res. Physiol. 2019, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ding, Y.; Dai, G.-L.; Zhang, Y.; Xu, M.-T.; Shen, J.-R.; Chen, T.-T.; Chen, Y.; Meng, G.-L. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 2021, 42, 230–241. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author | Treatment | Model | Number | Effect on Apoptosis | Effect on Fibrosis |
|---|---|---|---|---|---|
| Yang et al. [11] | empagliflozin | rats with cardiorenal syndrome | 18 | ↓ mitochondrial-Bax, cleaved caspase-3, and cleaved PARP | ↓ myocardial fibrosis |
| H9C2 cardiomyoblast cell line | 1.0 × 106 cells | ↓ apoptosis measured by flow cytometry | - | ||
| Yeh et al. [12] | sacubitril/valsartan | rats with cardiorenal syndrome | 24 | ↓ mitochondrial-Bax, cleaved caspase-3, and cleaved PARP | ↓ myocardial fibrosis |
| H9C2 cardiomyoblast cell line | 0.5 × 105 cells | ↓ apoptosis measured by flow cytometry | - | ||
| Liu et al. [13] | melatonin | mice on a high-fat diet | n.d. | ↓ apoptosis measured by TUNEL, ↑ anty-apoptotic Bcl-2 | no influence on fibrosis |
| H9C2 cardiomyoblast cell line | n.d. | medium with melatonin has a protective effect against apoptosis measured by TUNEL | - | ||
| Matyas et al. [14] | vardenafil | Zucker diabetic fatty rats | 30 | ↓ apoptosis measured by TUNEL and cleaved PARP | protective effects on myocardial fibrosis |
| Lin et al. [15] | irisin | mice with diabetic cardiomyopathy | n.d. | ↓ cleaved caspase-3 and ↑ the Bcl-2/Bax ratio | ↓ myocardial fibrosis |
| H9C2 cardiomyoblast cell line | n.d. | ↓ apoptosis measured by TUNEL and cleaved caspase-3 | - | ||
| neonatal rat cardiomyocyte isolation | n.d. | ↓ apoptosis measured by TUNEL and cleaved caspase-3 | - | ||
| Wang et al. [16] | physical activity | mice with diabetic cardiomyopathy | 24 | ↓ apoptosis measured by TUNEL and ↑ the Bcl-2/Bax ratio | ↓ myocardial fibrosis |
| First Author | Treatment | Model | Number | Effect on Ferroptosis | Effect on Fibrosis |
|---|---|---|---|---|---|
| Ma et al. [37] | canagliflozin | rats with a high-salt diet | 36 | ↓ ferroptosis: ↓ Fe2+, MDA, TFR1, ACSL4, 4-HNE, NOX4 and ↑ GSH, xCT, FTH1 | ↓ myocardial fibrosis |
| Kitakata et al. [38] | imeglimin | mice with a high-fat diet and L-NAME | n.d. | ↓ ferroptosis: ↑ GPX4 | ↓ myocardial fibrosis |
| Zhang et al. [39] | levosimendan | mice with a high-fat diet and L-NAME | n.d. | ↓ ferroptosis: ↑ xCT, GPX4, FSP-1, ↓ NOX4, total iron content, Fe2+, MDA, 4-HNE | not studied |
| Zhang et al. [40] | elabela | mice with angiotensin II-induced hypertension | n.d. | ↓ ferroptosis: ↑ GPX4, Nrf2, xCT | ↓ myocardial fibrosis |
| ferrostatin-1 | ↓ ferroptosis: ↑ GPX4, Nrf2, xCT | ↓ myocardial fibrosis | |||
| Su et al. [42] | ferrostatin-1 | mice with SIRT3 knockout (ferroptosis-dependent cardiac fibrosis) | n.d. | ↓ ferroptosis: ↓ 4-HNE level, ROS formation, TGF-β1 expression | ↓ myocardial fibrosis |
| First Author | Treatment | Model | Number | Type of PCD | Effect on PCD | Effect on Fibrosis |
|---|---|---|---|---|---|---|
| Xie et al. [49] | metformin | OVE26 mice with diabetic cardiomyopathy | 24 | autophagy | ↑ autophagy: measured by electron micrographic analysis of ventricular tissue double membrane-bound autophagic vesicles, ↑ LC3-II level, ↑ Beclin1 protein expression | not studied |
| apoptosis | ↓ apoptosis measured by TUNEL | |||||
| cardiomyocyte-derived cell line HL-1 | n.d. | autophagy | ↑ autophagy: ↑ LC3-II level | - | ||
| Xia et al. [60] | Spironolactone and rosuvastatin | HFpEF mice with metabolic disorders | n.d. | pyroptosis | ↓ pyroptosis: ↓ caspase-1, GSDMD, IL18 and IL-1β, NLRP3 expression in the EAT | not studied |
| autophagy | ↓ autophagy: ↓ LC3 level |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jankowski, J.; Kozub, K.O.; Kleibert, M.; Camlet, K.; Kleibert, K.; Cudnoch-Jędrzejewska, A. The Role of Programmed Types of Cell Death in Pathogenesis of Heart Failure with Preserved Ejection Fraction. Int. J. Mol. Sci. 2024, 25, 9921. https://doi.org/10.3390/ijms25189921
Jankowski J, Kozub KO, Kleibert M, Camlet K, Kleibert K, Cudnoch-Jędrzejewska A. The Role of Programmed Types of Cell Death in Pathogenesis of Heart Failure with Preserved Ejection Fraction. International Journal of Molecular Sciences. 2024; 25(18):9921. https://doi.org/10.3390/ijms25189921
Chicago/Turabian StyleJankowski, Jan, Kamil Oskar Kozub, Marcin Kleibert, Katarzyna Camlet, Klaudia Kleibert, and Agnieszka Cudnoch-Jędrzejewska. 2024. "The Role of Programmed Types of Cell Death in Pathogenesis of Heart Failure with Preserved Ejection Fraction" International Journal of Molecular Sciences 25, no. 18: 9921. https://doi.org/10.3390/ijms25189921