
Characterizing the Cell-Free Transcriptome in a Humanized Diffuse Large B-Cell Lymphoma Patient-Derived Tumor Xenograft Model for RNA-Based Liquid Biopsy in a Preclinical Setting

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. PDTX Growth, Therapy Response, and Toxicity
2.2. Blood Plasma Cell-Free RNA Concentration
2.3. Differential Abundance Analysis
2.3.1. Circulating Tumor cfRNA Profile
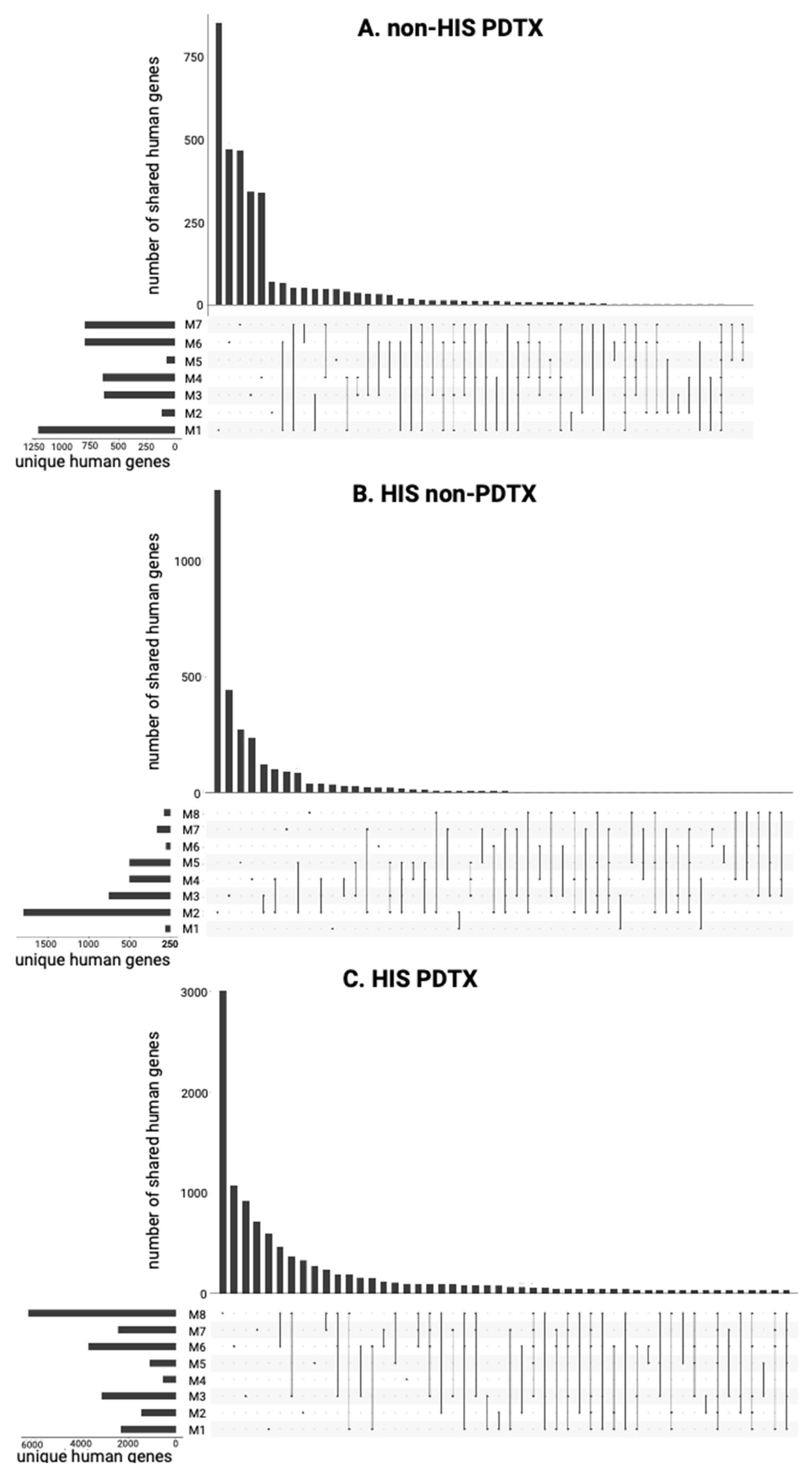
2.3.2. Variability in cfRNA Repertoire
2.4. Circulating Immune Cell Repertoire
2.4.1. Flow Cytometry
2.4.2. Computational Deconvolution Using cfRNA
3. Discussion
4. Materials and Methods
4.1. Humanized PDTX Model and Treatments
4.2. Sample Collection and Preparation
4.2.1. Murine Samples
4.2.2. Human Samples
4.3. Flow Cytometric Analysis
4.4. RNA Extraction
4.5. Library Preparation and Sequencing
4.6. Cell-Free RNA Concentration
4.7. Differential Abundance and Principal Component Analysis
4.8. Gene Set Enrichment Analysis
4.9. Deconvolution
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AFN | AcTaferons |
| cfDNA | cell-free DNA |
| cfRNA | cell-free RNA |
| CR | complete remission |
| ctRNA | circulating tumor RNA |
| DAG | differentially abundant gene |
| DLBCL | diffuse large B-cell lymphoma |
| EV | extracellular vesicle |
| FFPE | formalin-fixed paraffin-embedded tissue |
| GSEA | gene set enrichment analysis |
| JI | Jaccard Index |
| Log2FC | log2 fold change |
| mRNA | messenger RNA |
| mt-tRNA | mitochondrial transfer RNA |
| MYC | MYC proto-oncogene protein |
| PD | progressive disease |
| R-CHOP | rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisone |
| rRNA | ribosomal RNA |
| snRNA | small nuclear RNA |
| snoRNA | small nucleolar RNA |
| STAT3 | signal transducer and activator of transcription 3 |
| TNF | tumor necrosis factor |
| Wnt | wingless-related integration site |
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Müller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Davies, A.; Cummin, T.E.; Barrans, S.; Maishman, T.; Mamot, C.; Novak, U.; Caddy, J.; Stanton, L.; Kazmi-Stokes, S.; McMillan, A.; et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B cell lymphoma (REMoDL-B): An open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 649–662. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Chiappella, A.; Gascoyne, R.D.; Scott, D.W.; Zhang, Q.; Jurczak, W.; Özcan, M.; Hong, X.; Zhu, J.; Jin, J.; et al. ROBUST: A Phase III Study of Lenalidomide Plus R-CHOP Versus Placebo Plus R-CHOP in Previously Untreated Patients With ABC-Type Diffuse Large B cell Lymphoma. J. Clin. Oncol. 2021, 39, 1317–1328. [Google Scholar] [CrossRef]
- Younes, A.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Hong, X.; Zhu, J.; Patti, C.; Belada, D.; Samoilova, O.; Suh, C.; et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non–Germinal Center B cell Diffuse Large B cell Lymphoma. J. Clin. Oncol. 2019, 37, 1285–1295. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Author Correction: Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 1290–1291. [Google Scholar] [CrossRef]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.; Love, C.L.; Waldrop, A.; Leppä, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal Gene Signatures in Large-B cell Lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Monti, S. Molecular profiling of diffuse large B cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Ciavarella, S.; Vegliante, M.C.; Fabbri, M.; De Summa, S.; Melle, F.; Motta, G.; De Iuliis, V.; Opinto, G.; Enjuanes, A.; Rega, S.; et al. Dissection of DLBCL microenvironment provides a gene expression-based predictor of survival applicable to formalin-fixed paraffin-embedded tissue. Ann. Oncol. 2018, 29, 2363–2370. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Fan, N.; Tian, Y.; Tang, X.; Zhang, B.; Zhou, W.; Zhao, R.; Zhao, W. A Phase III, Randomized, Double-Blind, Placebo-Controlled, Multi-Center Study Evaluating the Efficacy and Safety of Orelabrutinib Plus R-CHOP Versus Placebo Plus R-CHOP in Treatment-Naïve Patients with Mcd Subtype Diffuse Large B cell Lymphoma. Blood 2022, 140 (Suppl. S1), 12110. [Google Scholar] [CrossRef]
- Zhang, M.-C.; Tian, S.; Fu, D.; Wang, L.; Cheng, S.; Yi, H.-M.; Jiang, X.-F.; Song, Q.; Zhao, Y.; He, Y.; et al. Genetic subtype-guided immunochemotherapy in diffuse large B cell lymphoma: The randomized GUIDANCE-01 trial. Cancer Cell 2023, 41, 1705–1716.e5. [Google Scholar] [CrossRef]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse large B cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef]
- Sole, C.; Arnaiz, E.; Manterola, L.; Otaegui, D.; Lawrie, C.H. The circulating transcriptome as a source of cancer liquid biopsy biomarkers. Semin. Cancer Biol. 2019, 58, 100–108. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Decruyenaere, P.; Offner, F.; Vandesompele, J. Circulating RNA biomarkers in diffuse large B cell lymphoma: A systematic review. Exp. Hematol. Oncol. 2021, 10, 13. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, W.; Qian, H.; Yin, Q.; Zhu, W.; Yan, Y. Circulating RNA as a novel tumor marker: An in vitro study of the origins and characteristics of extracellular RNA. Cancer Lett. 2007, 259, 50–60. [Google Scholar] [CrossRef]
- De Jong, O.G.; Verhaar, M.C.; Chen, Y.; Vader, P.; Gremmels, H.; Posthuma, G.; Schiffelers, R.M.; Gucek, M.; Van Balkom, B.W.M. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J. Extracell. Vesicles 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Bai, L.; Hong, M.; Ouyang, J.; Wang, R.; Zhang, X.; Chen, P. A Comprehensive Review on Circulating cfRNA in Plasma: Implications for Disease Diagnosis and Beyond. Diagnostics 2024, 14, 1045. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Daniel, V.C.; Marchionni, L.; Hierman, J.S.; Rhodes, J.T.; Devereux, W.L.; Rudin, C.M.; Yung, R.; Parmigiani, G.; Dorsch, M.; Peacock, C.D.; et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009, 69, 3364–3373. [Google Scholar] [CrossRef]
- Fichtner, I.; Rolff, J.; Soong, R.; Hoffmann, J.; Hammer, S.; Sommer, A.; Becker, M.; Merk, J. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin. Cancer Res. 2008, 14, 6456–6468. [Google Scholar] [CrossRef]
- Wang, C.-F.; Shi, X.-J. Generation and application of patient-derived xenograft models in pancreatic cancer research. Chin. Med. J. 2019, 132, 2729–2736. [Google Scholar] [CrossRef]
- Malaney, P.; Nicosia, S.V.; Davé, V. One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Lett. 2013, 344, 1–12. [Google Scholar] [CrossRef]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Establishment of a Patient-Derived Tumor Xenograft Model and Application for Precision Cancer Medicine. Chem. Pharm. Bull. 2018, 66, 225–230. [Google Scholar] [CrossRef]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Cauwels, A.; Pieters, T.; De Smedt, R.; T’sas, S.; Almeida, A.; Daneels, W.; Van Vlierberghe, P.; Tavernier, J. Direct and indirect anti-leukemic properties of activity-on-target interferons for the treatment of T cell acute lymphoblastic leukemia. Haematologica 2021, 107, 1448–1453. [Google Scholar] [CrossRef] [PubMed]
- Daneels, W.; Van Parys, A.; Huyghe, L.; Rogge, E.; De Rouck, S.; Christiaen, R.; Zabeau, L.; Taveirne, S.; Van Dorpe, J.; Kley, N.; et al. High efficacy of huCD20-targeted AcTaferon in humanized patient derived xenograft models of aggressive B cell lymphoma. Exp. Hematol. Oncol. 2024, 13, 59. [Google Scholar] [CrossRef]
- Hodkinson, B.P.; Schaffer, M.; Brody, J.D.; Jurczak, W.; Carpio, C.; Ben-Yehuda, D.; Avivi, I.; Forslund, A.; Özcan, M.; Alvarez, J.; et al. Biomarkers of response to ibrutinib plus nivolumab in relapsed diffuse large B cell lymphoma, follicular lymphoma, or Richter’s transformation. Transl. Oncol. 2020, 14, 100977. [Google Scholar] [CrossRef]
- Monti, S.; Chapuy, B.; Takeyama, K.; Rodig, S.J.; Hao, Y.; Yeda, K.T.; Inguilizian, H.; Mermel, C.; Currie, T.; Dogan, A.; et al. Integrative Analysis Reveals an Outcome-Associated and Targetable Pattern of p53 and Cell Cycle Deregulation in Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 22, 359–372. [Google Scholar] [CrossRef]
- Xu, Q.; Tan, C.; Ni, S.; Wang, Q.; Wu, F.; Liu, F.; Ye, X.; Meng, X.; Sheng, W.; Du, X. Identification and validation of a two-gene expression index for subtype classification and prognosis in Diffuse Large B cell Lymphoma. Sci. Rep. 2015, 5, 10006. [Google Scholar] [CrossRef]
- Chen, X.; Lu, T.; Cai, Y.; Han, Y.; Ding, M.; Chu, Y.; Zhou, X.; Wang, X. KIAA1429-mediated m6A modification of CHST11 promotes progression of diffuse large B cell lymphoma by regulating Hippo–YAP pathway. Cell. Mol. Biol. Lett. 2023, 28, 32. [Google Scholar] [CrossRef]
- Hu, S.; Ren, S.; Cai, Y.; Liu, J.; Han, Y.; Zhao, Y.; Yang, J.; Zhou, X.; Wang, X. Glycoprotein PTGDS promotes tumorigenesis of diffuse large B cell lymphoma by MYH9-mediated regulation of Wnt–β-catenin–STAT3 signaling. Cell Death Differ. 2021, 29, 642–656. [Google Scholar] [CrossRef]
- Tian, T.; Li, J.; Shi, D.; Zeng, Y.; Yu, B.; Li, X.; Wei, P.; Zhou, X. SMYD3 promotes aerobic glycolysis in diffuse large B cell lymphoma via H3K4me3-mediated PKM2 transcription. Cell Death Dis. 2022, 13, 763. [Google Scholar] [CrossRef]
- Carey, V. Enhancements HRfCl (). ROC: Utilities for ROC, with Microarray Focus. R Package Version 1.80.0. Available online: https://www.bioconductor.org (accessed on 10 January 2024).
- Roskams-Hieter, B.; Kim, H.J.; Anur, P.; Wagner, J.T.; Callahan, R.; Spiliotopoulos, E.; Kirschbaum, C.W.; Civitci, F.; Spellman, P.T.; Thompson, R.F.; et al. Plasma cell-free RNA profiling distinguishes cancers from pre-malignant conditions in solid and hematologic malignancies. NPJ Precis. Oncol. 2022, 6, 28. [Google Scholar] [CrossRef]
- Zhuang, J.; Ibarra, A.; Acosta, A.; Karns, A.P.; Aballi, J.; Nerenberg, M.; Sninsky, J.J.; Quake, S.R.; Toden, S. Survey of extracellular communication of systemic and organ-specific inflammatory responses through cell free messenger RNA profiling in mice. EBioMedicine 2022, 83, 104242. [Google Scholar] [CrossRef] [PubMed]
- Vermeirssen, V.; Deleu, J.; Morlion, A.; Everaert, C.; De Wilde, J.; Anckaert, J.; Durinck, K.; Nuytens, J.; Rishfi, M.; Speleman, F.; et al. Whole transcriptome profiling of liquid biopsies from tumour xenografted mouse models enables specific monitoring of tumour-derived extracellular RNA. NAR Cancer 2022, 4, zcac037. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Zhuang, J.; Zhao, Y.; Salathia, N.S.; Huang, V.; Acosta, A.D.; Aballi, J.; Toden, S.; Karns, A.P.; Purnajo, I.; et al. Non-invasive characterization of human bone marrow stimulation and reconstitution by cell-free messenger RNA sequencing. Nat. Commun. 2020, 11, 400. [Google Scholar] [CrossRef]
- Toden, S.; Zhuang, J.; Acosta, A.D.; Karns, A.P.; Salathia, N.S.; Brewer, J.B.; Nerenberg, M.; Quake, S.R.; Ibarra, A. Non-invasive characterization of Alzheimer’s disease by circulating, cell-free messenger RNA next-generation sequencing. Sci. Adv. 2020, 6, eabb1654. [Google Scholar] [CrossRef]
- Everaert, C.; Helsmoortel, H.; Decock, A.; Hulstaert, E.; Van Paemel, R.; Verniers, K.; Nuytens, J.; Anckaert, J.; Nijs, N.; Tulkens, J.; et al. Performance assessment of total RNA sequencing of human biofluids and extracellular vesicles. Sci. Rep. 2019, 9, 17547. [Google Scholar] [CrossRef]
- Eskandari, M.; Manoochehrabadi, S.; Pashaiefar, H.; Zaimy, M.A.; Ahmadvand, M. Clinical significance of cell-free DNA as a prognostic biomarker in patients with diffuse large B cell lymphoma. Blood Res. 2019, 54, 114–119. [Google Scholar] [CrossRef]
- Shirouchi, Y.; Mishima, Y.; Takayama, T.; Minowa, S.; Ishihara, Y.; Tamba, M.; Hirano, M.; Onda, N.; Takeuchi, K.; Maruyama, D. Serum cell-free DNA concentration as a possible prognostic marker in newly diagnosed diffuse large B cell lymphoma. Biomed. Res. 2022, 43, 99–106. [Google Scholar] [CrossRef]
- Decruyenaere, P.; Giuili, E.; Verniers, K.; Anckaert, J.; De Grove, K.; Van der Linden, M.; Deeren, D.; Van Dorpe, J.; Offner, F.; Vandesompele, J. Exploring the cell-free total RNA transcriptome in diffuse large B cell lymphoma and primary mediastinal B cell lymphoma patients as biomarker source in blood plasma liquid biopsies. Front. Oncol. 2023, 13, 1221471. [Google Scholar] [CrossRef]
- Deleu, J.; Van Droogenbroeck, H.; Anckaert, J.; Decock, A.; De Wilde, J.; Durinck, K.; Mus, L.M.; Nuytens, J.; Rishfi, M.; Schoofs, K.; et al. Exploration of neuroblastoma xenograft models for tumor extracellular RNA profiling in murine blood plasma. ExRNA 2023, 5, 0007. [Google Scholar] [CrossRef]
- Van Goethem, A.; Deleu, J.; Yigit, N.; Everaert, C.; Moreno-Smith, M.; Vasudevan, S.A.; Zeka, F.; Demuynck, F.; Barbieri, E.; Speleman, F.; et al. Longitudinal evaluation of serum microRNAs as biomarkers for neuroblastoma burden and therapeutic p53 reactivation. NAR Cancer 2023, 5, zcad002. [Google Scholar] [CrossRef]
- Zeka, F.; Decock, A.; Van Goethem, A.; Vanderheyden, K.; Demuynck, F.; Lammens, T.; Helsmoortel, H.H.; Vermeulen, J.; Noguera, R.; Berbegall, A.P.; et al. Circulating microRNA biomarkers for metastatic disease in neuroblastoma patients. J. Clin. Investig. 2018, 3, e97021. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, A.; Vanderburg, C.; McDonald, J.T.; Ramkumar, C.; Kadungure, T.; Zhang, H.; Gartenhaus, R.B.; Evens, A.M. A Circulating microRNA Signature Predicts Age-Based Development of Lymphoma. PLoS ONE 2017, 12, e0170521. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, A.; Stevenson, K.; Vanderburg, C.; Ravi, D.; McDonald, J.T.; Christie, A.L.; Shigemori, K.; Jester, H.; Weinstock, D.M. Identification of Circulating Serum Multi-MicroRNA Signatures in Human DLBCL Models. Sci. Rep. 2019, 9, 17161. [Google Scholar] [CrossRef]
- Rinaldi, F.; Marchesi, F.; Palombi, F.; Pelosi, A.; Di Pace, A.L.; Sacconi, A.; Terrenato, I.; Annibali, O.; Tomarchio, V.; Marino, M.; et al. MiR-22, a serum predictor of poor outcome and therapy response in diffuse large B cell lymphoma patients. Br. J. Haematol. 2021, 195, 399–404. [Google Scholar] [CrossRef]
- Baldasici, O.; Balacescu, L.; Cruceriu, D.; Roman, A.; Lisencu, C.; Fetica, B.; Visan, S.; Cismaru, A.; Jurj, A.; Barbu-Tudoran, L.; et al. Circulating Small EVs miRNAs as Predictors of Pathological Response to Neo-Adjuvant Therapy in Breast Cancer Patients. Int. J. Mol. Sci. 2022, 23, 12625. [Google Scholar] [CrossRef]
- Khare, D.; Goldschmidt, N.; Bardugo, A.; Gur-Wahnon, D.; Ben-Dov, I.Z.; Avni, B. Plasma microRNA profiling: Exploring better biomarkers for lymphoma surveillance. PLoS ONE 2017, 12, e0187722. [Google Scholar] [CrossRef]
- Cui, Q.; Vari, F.; Cristino, A.S.; Salomon, C.; Rice, G.E.; Sabdia, M.B.; Guanzon, D.; Palma, C.; Mathew, M.; Talaulikar, D.; et al. Circulating cell-free miR-494 and miR-21 are disease response biomarkers associated with interim-positron emission tomography response in patients with diffuse large B cell lymphoma. Oncotarget 2018, 9, 34644–34657. [Google Scholar] [CrossRef]
- Inada, K.; Okoshi, Y.; Cho, Y.; Saito, H.; Iijima, T.; Hori, M.; Kojima, H. Availability of Circulating MicroRNAs as a Biomarker for Early Diagnosis of Diffuse Large B cell Lymphoma. Open J. Blood Dis. 2015, 5, 48–58. [Google Scholar] [CrossRef]
- Di, C.; Jiang, Y.; Li, M.; Juan, X.; Xu, C. Circulating Exosomal microRNA Signature As a Non-invasive Biomarker for Diagnosis of Diffuse Large B cell Lymphoma. Blood 2018, 132, 5406. [Google Scholar] [CrossRef]
- Zare, N.; Javanmard, S.H.; Mehrzad, V.; Eskandari, N.; Kefayat, A. Evaluation of exosomal miR-155, let-7g and let-7i levels as a potential non-invasive biomarker among refractory/relapsed patients, responsive patients and patients receiving R-CHOP. Leuk. Lymphoma 2019, 60, 1877–1889. [Google Scholar] [CrossRef]
- Morlion, A.; Decruyenaere, P.; Schoofs, K.; Anckaert, J.; Nuytens, J.; Vanden, E.E.; Verniers, K.; Everaert, C.; Offner, F.; Van Dorpe, J.; et al. Patient-Specific Alterations in Blood Plasma cfRNA Profiles Enable Accurate Classification of Cancer Patients and Controls. medRxiv 2023. Available online: http://medrxiv.org/content/early/2023/05/28/2023.05.24.23290388.abstract (accessed on 10 January 2024).
- Williams, A.L.; Fitzgerald, J.E.; Ivich, F.; Sontag, E.D.; Niedre, M. Short-Term Circulating Tumor Cell Dynamics in Mouse Xenograft Models and Implications for Liquid Biopsy. Front. Oncol. 2020, 10, 601085. [Google Scholar] [CrossRef]
- Jin, Z.; Xiang, R.; Qing, K.; Li, D.; Liu, Z.; Li, X.; Zhu, H.; Zhang, Y.; Wang, L.; Xue, K.; et al. Lenalidomide overcomes the resistance to third-generation CD19-CAR-T cell therapy in preclinical models of diffuse large B cell lymphoma. Cell. Oncol. 2023, 46, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Wu, J.-C.; Liu, P.; Li, Z.-J.; Wang, Y.; Chen, B.-Y.; Hu, C.-L.; Fei, M.-Y.; Yu, P.-C.; Jiang, Y.-L.; et al. Inhibition of USP1 reverses the chemotherapy resistance through destabilization of MAX in the relapsed/refractory B cell lymphoma. Leukemia 2023, 37, 164–177. [Google Scholar] [CrossRef]
- Goodstal, S.M.; Lin, J.; Crandall, T.; Crowley, L.; Bender, A.T.; Pereira, A.; Soloviev, M.; Wesolowski, J.S.; Iadevaia, R.; Schelhorn, S.-E.; et al. Preclinical evidence for the effective use of TL-895, a highly selective and potent second-generation BTK inhibitor, for the treatment of B cell malignancies. Sci. Rep. 2023, 13, 20412. [Google Scholar] [CrossRef]
- Koh, W.; Pan, W.; Gawad, C.; Fan, H.C.; Kerchner, G.A.; Wyss-Coray, T.; Blumenfeld, Y.J.; El-Sayed, Y.Y.; Quake, S.R. Non-invasive in vivo monitoring of tissue-specific global gene expression in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7361–7366. [Google Scholar] [CrossRef]
- Wagner, J.T.; Kim, H.J.; Johnson-Camacho, K.C.; Kelley, T.; Newell, L.F.; Spellman, P.T.; Ngo, T.T.M. Diurnal stability of cell-free DNA and cell-free RNA in human plasma samples. Sci. Rep. 2020, 10, 16456. [Google Scholar] [CrossRef]
- Cabús, L.; Lagarde, J.; Curado, J.; Lizano, E.; Pérez-Boza, J. Current challenges and best practices for cell-free long RNA biomarker discovery. Biomark. Res. 2022, 10, 62. [Google Scholar] [CrossRef]
- Consortium, T.; Anckaert, J.; Cobos, F.A.; Decock, A.; Decruyenaere, P.; Deleu, J.; De Preter, K.; De Wever, O.; De Wilde, J.; Dhondt, B.; et al. Performance Evaluation of RNA Purification Kits and Blood Collection Tubes in the Extracellular RNA Quality Control (exRNAQC) study. bioRxiv 2022. Available online: http://biorxiv.org/content/early/2022/12/27/2021.05.11.442610.abstract (accessed on 10 January 2024).
- Van Der Schueren, C.; Decruyenaere, P.; Cobos, F.A.; Bult, J.; Deleu, J.; Dipalo, L.L.; Helsmoortel, H.H.; Hulstaert, E.; Morlion, A.; Varas, E.R.; et al. Subpar reporting of pre-analytical variables in RNA—Focused blood plasma studies. Mol. Oncol. 2024. [Google Scholar] [CrossRef]
- Brinkman, K.; Meyer, L.; Bickel, A.; Enderle, D.; Berking, C.; Skog, J.; Noerholm, M. Extracellular vesicles from plasma have higher tumour RNA fraction than platelets. J. Extracell. Vesicles 2020, 9, 1741176. [Google Scholar] [CrossRef] [PubMed]
- Mian, S.A.; Anjos-Afonso, F.; Bonnet, D. Advances in Human Immune System Mouse Models for Studying Human Hematopoiesis and Cancer Immunotherapy. Front. Immunol. 2021, 11, 619236. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2014. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 January 2024).
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Neph, S.; Kuehn, M.S.; Reynolds, A.P.; Haugen, E.; Thurman, R.E.; Johnson, A.K.; Rynes, E.; Maurano, M.T.; Vierstra, J.; Thomas, S.; et al. BEDOPS: High-performance genomic feature operations. Bioinformatics 2012, 28, 1919–1920. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RseQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Hulstaert, E.; Decock, A.; Morlion, A.; Everaert, C.; Verniers, K.; Nuytens, J.; Nijs, N.; Schroth, G.P.; Kuersten, S.; Gross, S.M.; et al. Protocol Messenger RNA capture sequencing of extracellular RNA from human biofluids using a comprehensive set of spike-in controls. STAR Protoc. 2021, 2, 100475. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2018. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 10 January 2024).
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.S.; Theis, F.J.; Yosef, N. Impulse model-based differential expression analysis of time course sequencing data. Nucleic Acids Res. 2018, 46, e119. [Google Scholar] [CrossRef] [PubMed]
- Marini, F.; Binder, H. pcaExplorer: An R/Bioconductor package for interacting with RNA-seq principal components. BMC Bioinform. 2019, 20, 331. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol. 2019, 20, 1. [Google Scholar] [CrossRef]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef]
- Yu, G. enrichplot: Visualization of Functional Enrichment Result. R Package Version 1.24.0. 2024. Available online: https://yulab-smu.top/biomedical-knowledge-mining-book/ (accessed on 10 January 2024).
- Vorperian, S.K.; Moufarrej, M.N.; Quake, S.R. Cell types of origin of the cell-free transcriptome. Nat. Biotechnol. 2022, 40, 855–861. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.R-project.org/ (accessed on 14 January 2024).







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Decruyenaere, P.; Daneels, W.; Morlion, A.; Verniers, K.; Anckaert, J.; Tavernier, J.; Offner, F.; Vandesompele, J. Characterizing the Cell-Free Transcriptome in a Humanized Diffuse Large B-Cell Lymphoma Patient-Derived Tumor Xenograft Model for RNA-Based Liquid Biopsy in a Preclinical Setting. Int. J. Mol. Sci. 2024, 25, 9982. https://doi.org/10.3390/ijms25189982
Decruyenaere P, Daneels W, Morlion A, Verniers K, Anckaert J, Tavernier J, Offner F, Vandesompele J. Characterizing the Cell-Free Transcriptome in a Humanized Diffuse Large B-Cell Lymphoma Patient-Derived Tumor Xenograft Model for RNA-Based Liquid Biopsy in a Preclinical Setting. International Journal of Molecular Sciences. 2024; 25(18):9982. https://doi.org/10.3390/ijms25189982
Chicago/Turabian StyleDecruyenaere, Philippe, Willem Daneels, Annelien Morlion, Kimberly Verniers, Jasper Anckaert, Jan Tavernier, Fritz Offner, and Jo Vandesompele. 2024. "Characterizing the Cell-Free Transcriptome in a Humanized Diffuse Large B-Cell Lymphoma Patient-Derived Tumor Xenograft Model for RNA-Based Liquid Biopsy in a Preclinical Setting" International Journal of Molecular Sciences 25, no. 18: 9982. https://doi.org/10.3390/ijms25189982
APA StyleDecruyenaere, P., Daneels, W., Morlion, A., Verniers, K., Anckaert, J., Tavernier, J., Offner, F., & Vandesompele, J. (2024). Characterizing the Cell-Free Transcriptome in a Humanized Diffuse Large B-Cell Lymphoma Patient-Derived Tumor Xenograft Model for RNA-Based Liquid Biopsy in a Preclinical Setting. International Journal of Molecular Sciences, 25(18), 9982. https://doi.org/10.3390/ijms25189982