Abstract
Tetraallylsilane can undergo either a mono or double rearrangement when treated with iodine (I2). The extent of rearrangement depends on the equivalents of I2 used, where 1 equivalent gives high yields of mono-rearranged products and excess (e.g., 3 equivalents) causes double rearrangement to occur. This transformation can be applied to the synthesis of potentially valuable silicon-stereogenic organosilanes.
1. Introduction
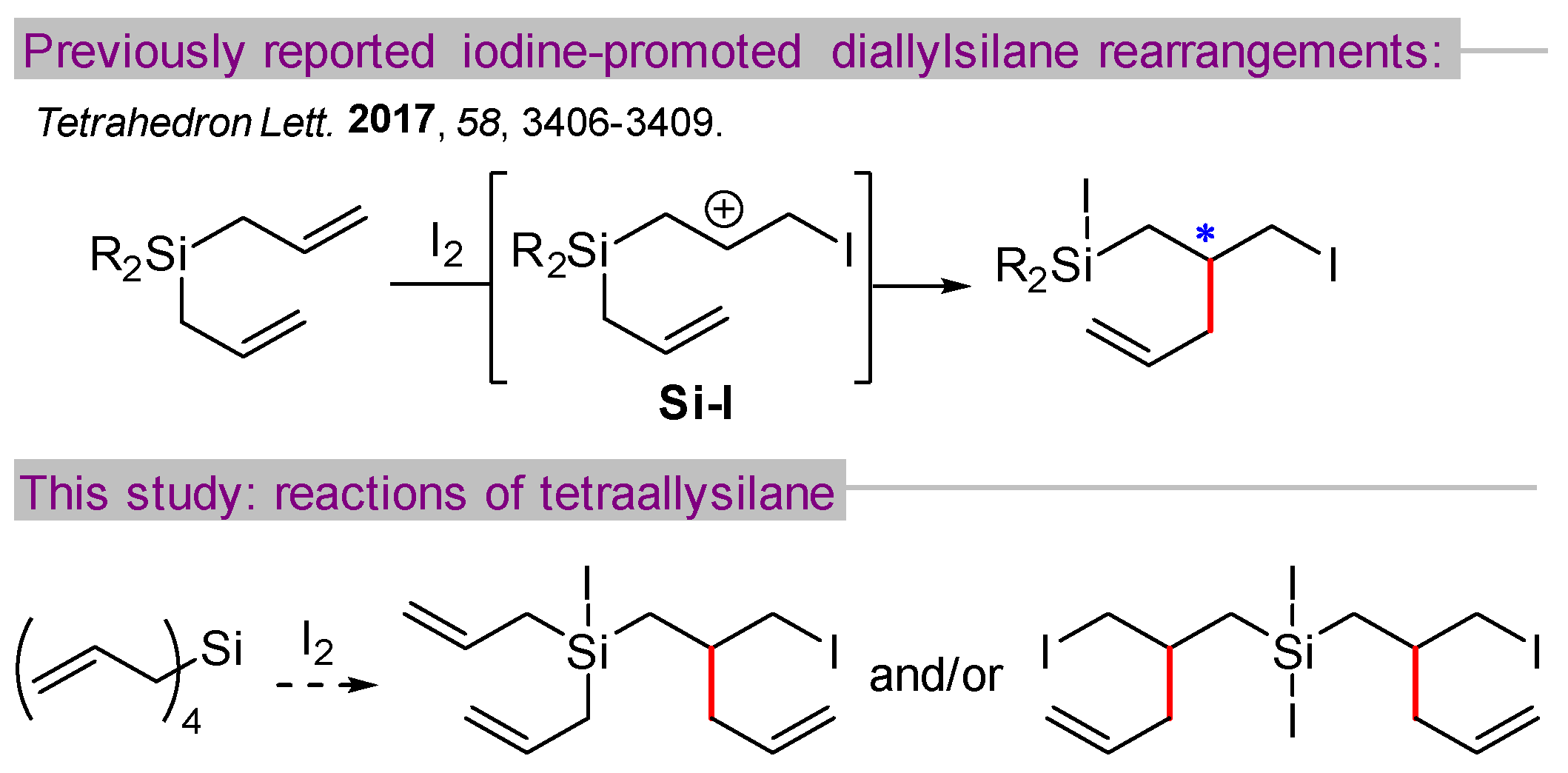
During the course of investigating iodine-catalyzed etherification reactions of allylsilanes, our group discovered an iodine-promoted rearrangement of diallylsilanes (Scheme 1) [1]. The reaction is thought to proceed through a mechanism involving the intramolecular allylation of a beta-silyl carbocation Si-I, generating a new carbon–carbon bond (highlighted in red) and a new stereocenter (*). Several different diallylsilanes with differing substitutions at silicon (i.e., Me2, i-Pr2, Ph2, cyclotrimethylene) were effectively engaged in this transformation, with diallyldiphenylsilane giving the best ratio of rearrangement versus competing iododeallylation.
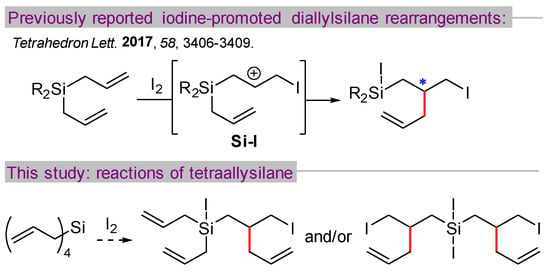
Scheme 1.
Iodine-promoted rearrangement of diallylsilanes [1].
We questioned how tetraallylsilane might behave in this reaction, presenting not only an opportunity for a double rearrangement process but also perhaps an increased likelihood of competing side reactions (e.g., deallylation [2] or polymerization [3]). In this paper, we report results from reactions of tetraallylsilane with iodine, demonstrating the ability to select for mono- or double-rearrangement products in good yield. Additionally, this strategy was used to generate potentially valuable silacyclic compounds containing a stereogenic silicon atom.
2. Results and Discussion
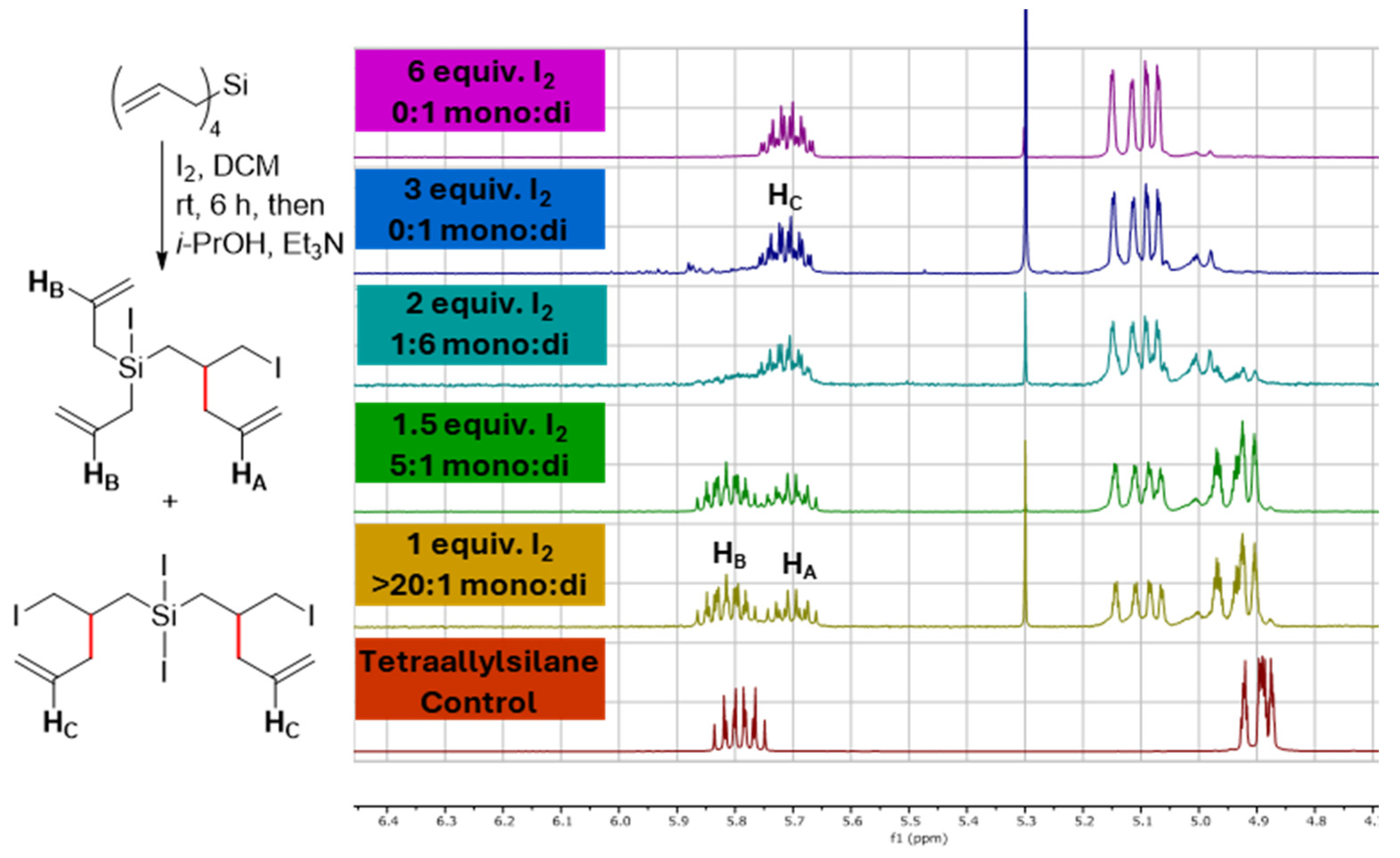
To begin our investigation, tetraallylsilane was treated with different equivalents of I2 using CD2Cl2 as the solvent, allowing for direct product analysis with NMR (Figure 1). As shown in Figure 1, using 1.0 equiv. of I2 resulted in the nearly complete consumption of the starting material and the selective formation of the mono-rearranged product. Increasing the amount of I2 to 2 equivalents then caused the di-rearranged product to predominate.
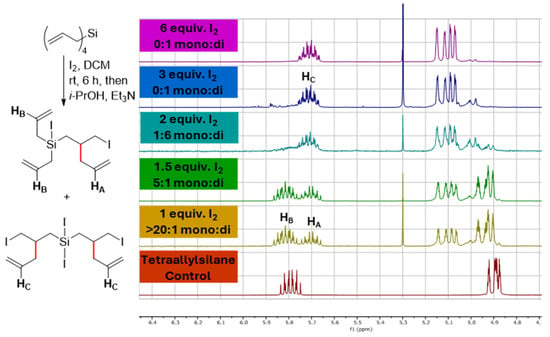
Figure 1.
NMR analysis of tetraallylsilane reactions with differing equivalents of iodine (I2) showing that good selectivity for the single (mono) and double (di)-rearranged products could be obtained by controlling the amount of I2 used.
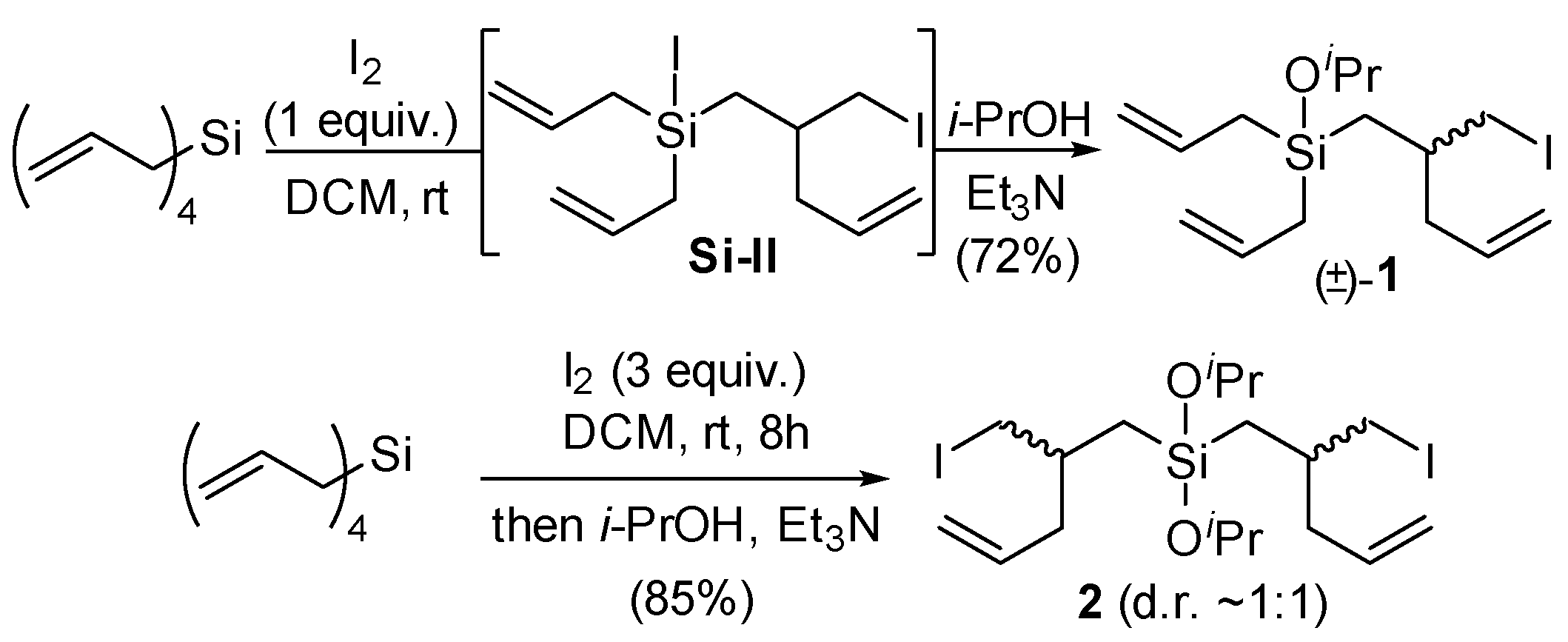
From the equivalents study, 1.0 equiv. of I2 was selected as optimal for the mono rearrangement of tetraallylsilane, and 3.0 equiv. of I2 was selected for the double rearrangement. In this way, compounds 1 and 2 were obtained in 72% and 85% yields, respectively (Scheme 2). The high yield of 1 (i.e., >50%) obtained when using stoichiometric I2 is noteworthy, as a statistical mixture of tetraallylsilane, 1, and 2 may have been anticipated. Indeed, compared with other reported methods for diallylsilane rearrangements (e.g., acid-promoted [4,5]), only the use of iodine was found to provide good yields of mono- vs. di-rearranged products with limited byproduct formation (e.g., deallylation) in reactions with tetraallylsilane. We attribute the selectivity in part to the reduced nucleophilicity of the initially formed iodosilane intermediate Si-I, allowing for the selective consumption of tetraallylsilane over Si-I.
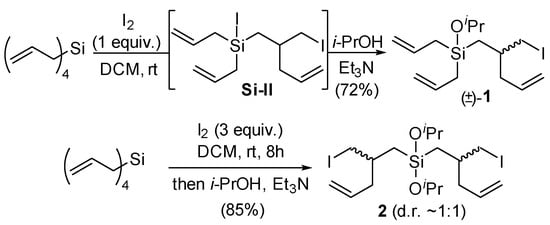
Scheme 2.
Synthesis of 1 and 2 from tetraallylsilane by iodine-promoted rearrangement.
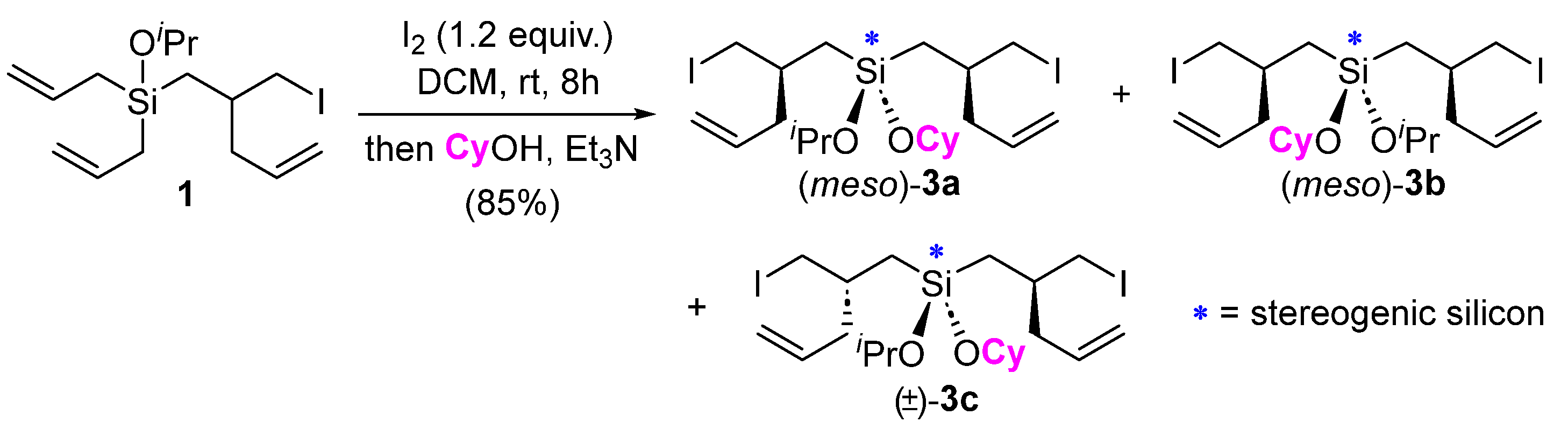
Compound 1, being a diallylsilane, could also be reengaged in an iodine-promoted rearrangement. Performing this type of sequential rearrangement allows for the incorporation of different alcohols for subsequent etherification. For instance, treatment of 1 with I2 followed by cyclohexanol gave compound 3, featuring two different alkoxy ligands attached to silicon (Scheme 3). This strategy might prove useful for reactions involving two different silicon-tethered alcohols where the use of a dichlorosilane for this purpose is challenging (e.g., due to homocoupling) [6,7]. There are three possible diastereomers of compound 3 (i.e., 3a–c) that we were unable to separate with flash column chromatography on silica. The 1H NMR signals for these diastereomers were overlapping and/or coincident; however, some signals were resolved by 13C NMR, making it clear that all three diastereomers had been obtained (see Supporting Information).
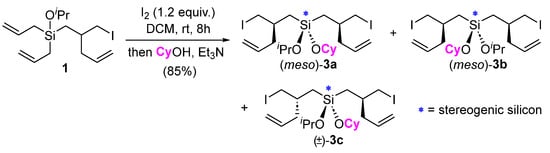
Scheme 3.
Further reaction of iodine rearrangement product 1 and the generation of 3, featuring two different alkoxy ligands on silicon. All three diastereomers contain a stereogenic silicon atom, according to the IUPAC definition, where the interchange of any two groups leads to a stereoisomer. For instance, interchanging the alkoxy groups in 3a leads to its diastereomer 3b, and a switch of the alkoxy groups on 3c produces its enantiomer.
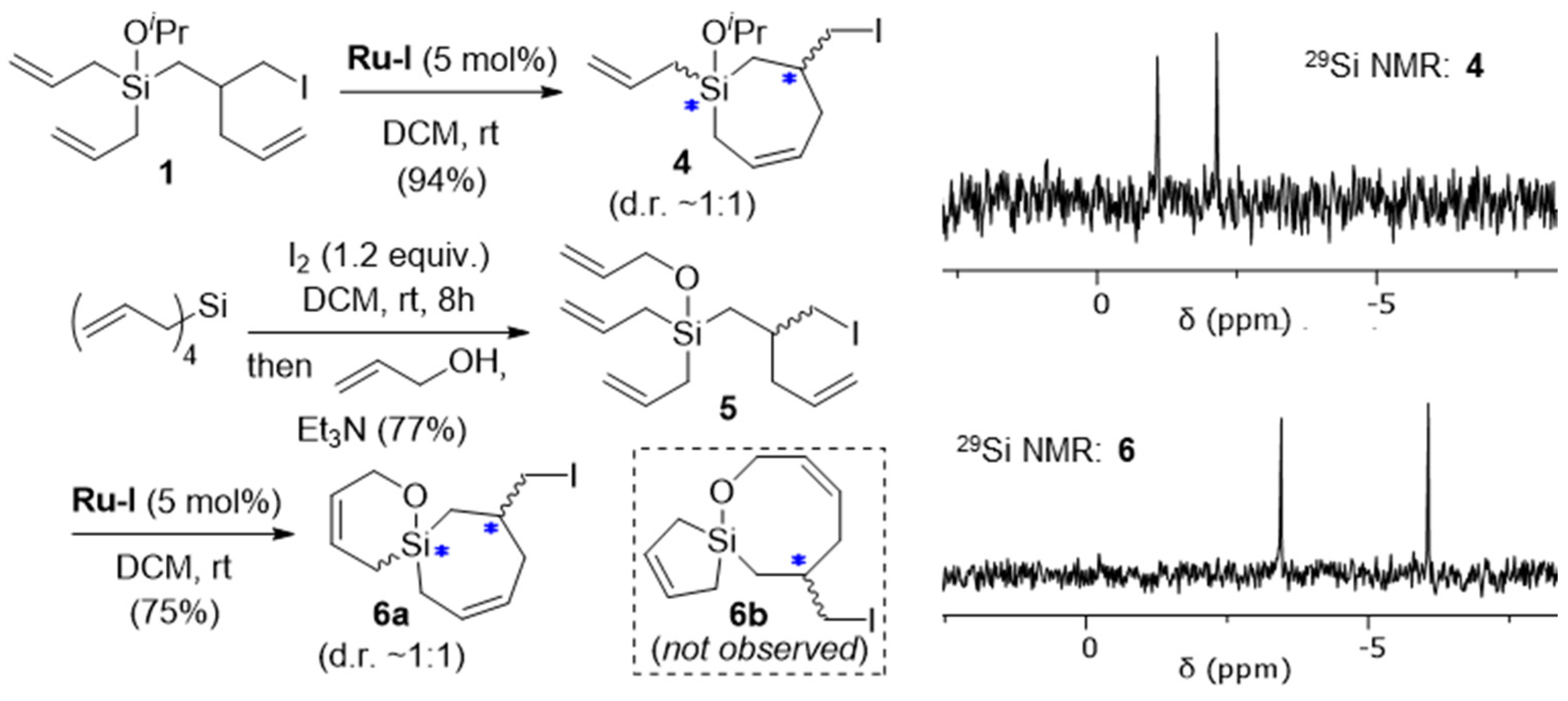
Each of the diasteromers 3a–c contain a stereogenic silicon atom, according to the IUPAC definition, where the interchange of any two groups attached to silicon leads to a stereoisomer (e.g., from 3a to 3b and vice versa) [8]. Compared with the plethora of methods available for the asymmetric synthesis of purely organic molecules, synthetic approaches to silicon-stereogenic organosilanes remain underdeveloped [9,10,11,12,13,14,15,16] despite the potential value of these compounds in biology and material science [17,18,19]. We were inspired, therefore, to consider how this method might be further employed to generate structures containing stereogenic silicon atoms. Building off our previous success with ring-closing metathesis (RCM) reactions of iodine-mediated diallylsilane rearrangement products [20], RCM was pursued as a route to potentially differentiate the olefinic ligands attached to silicon in our rearranged products. In the event, the treatment of a dilute (0.02M) solution of 1 in DCM with Grubbs’ first-generation catalyst (Ru-I) at room temperature resulted in its clean conversion to the seven-membered ring silicon-stereogenic silacycle 4 in a high yield (Scheme 4). No evidence for the potential silacyclopentene product was observed with NMR, consistent with Ahmad and coworkers’ observation that silacycloheptene formation is easier than silacyclopentene with RCM [21]. Given the thermodynamic nature of RCM, the results suggests that the ring strain of the seven-membered silacycloalkene is less than that of the five-membered silacycloalkene. Consistent with these results and that of Mahieux et al., who observed selective six-membered ring formation over five-membered ring formation when studying silacycle formation with RCM [22], the RCM of 5 selectively produced the 6,7-spirocycle 6a rather than the alternative 5,8-spirocyclic isomer 6b. Compound 6a was isolated in a 75% yield, with the mass balance appearing to be products arising from cross-metathesis/polymerization, which might also explain the lack of 6b in the product mixture as this less stable ring system may be susceptible to ring-opening metathesis processes [23,24,25,26]. The 29Si NMR spectra of 4 and 6 showed two signals of roughly equal intensity, indicating that a ~1:1 mixture of diastereomers was produced. The presence of two signals in the 29Si NMR also provides additional confirmation of their structure and differentiation from the alternative silacyclopentene RCM products (e.g., 6b), which do not contain a stereogenic silicon atom and would therefore only exhibit a single 29Si NMR signal (i.e., a mixture of enantiomers rather than diastereomers would have been produced). Moving forward, different approaches to controlling the relative and absolute stereochemistry of silicon-stereogenic compounds of type 3, 4, and 6 will be explored.
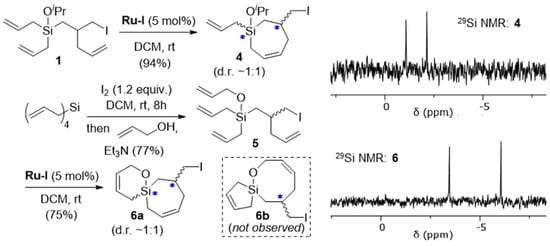
Scheme 4.
Synthesis of silacycle 4 and oxasilaspirocycle 6a containing stereogenic carbon and silicon atoms (*) with RCM. 29Si NMR data indicate these compounds were obtained as ~1:1 mixtures of diastereomers. The presence of two 29Si NMR signals also distinguishes 4 and 6a from other possible silacyclopentene RCM products (e.g., 6b).
3. Materials and Methods
3.1. General Information
All reactions were carried out in dry vessels under a nitrogen atmosphere at ambient conditions unless otherwise specified. The dry solvents used were prepared by passing the purchased solvents (Thermo Fisher Scientific, Rockford, IL, USA) through a column of activated alumina under nitrogen immediately prior to use. All reagents were purchased from Sigma Aldrich (St. Louis, MI, USA) and used as received unless mentioned otherwise. TLC analysis used 0.25 mm silica layer fluorescence UV254 plates (Silicycle, Quebec City, QC, CA). Column chromatography used silica gel (230–400 mesh; Silicycle, Quebec City, QC, CA). For IR, we used a Thermo Fisher Nicolet is10 FT-IR spectrometer equipped with a single-bounce diamond ATR. NMR spectra were recorded on a Bruker 500 MHz spectrometer in CDCl3; chemical shifts (d) are given in ppm, and coupling constants (J) are given in Hz. Solvent signals were used as references (CDCl3: δc = 77.0 ppm; residual CHCl3 in CDCl3: δH = 7.26 ppm). For HRMS, we used a Bruker Maxis Impact quadrupole time-of-flight LC–MS with electrospray ionization (ESI positive).
3.2. Compound Syntheses and Spectral Data
Diallyl(2-(iodomethyl)pent-4-en-1-yl)(isopropoxy)silane (1). To a solution of tetraallylsilane (0.19 g, 1.0 mmol) in DCM (10 mL), I2 (0.25 g, 1.0 mmol) was added, and the mixture was stirred for 6 h. The solution was then cooled to 0 °C before triethylamine (2.0 mmol) and isopropanol (0.13 mL, 1.5 mmol) were added, and the resulting mixture was stirred and allowed to slowly warm to room temperature over 6 h. The reaction was quenched with water (15 mL) and extracted with DCM (2 × 15 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated on a rotary evaporator. The crude product was purified with column chromatography on silica (20:1 to 10:1 Hexanes:EtOAc), giving 1 (0.27 g, 72%) as an oil. IR (ATR) 3069, 2970, 2911, 1639, 1589, 1428, 1381, 1368, 1219, 1109, 1021, 997, 914, 877, 732 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.89–5.76 (m, 2H), 5.70 (ddt, J = 17.2, 10.1, 7.2 Hz, 1H), 5.13 (dd, J = 17.2, 1.8 Hz, 1H), 5.10–5.06 (m, 1H), 4.98–4.89 (m, 4H), 4.08 (dt, J = 12.1, 6.0 Hz, 1H), 3.36 (dd, J = 9.5, 4.3 Hz, 1H), 3.28 (dd, J = 9.5, 5.4 Hz, 1H), 2.21 (dddt, J = 13.7, 6.8, 5.5, 1.4 Hz, 1H), 2.16–2.03 (m, 1H), 1.75–1.65 (m, 3H), 1.59 (dddd, J = 12.6, 6.9, 3.5, 1.4 Hz, 1H), 1.33–1.23 (m, 1H), 1.16 (d, J = 6.0 Hz, 7H), 0.71 (dd, J = 6.8, 4.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 135.61, 133.49, 117.29, 114.46, 114.44, 65.55, 41.04, 34.68, 30.90, 25.83, 22.47, 22.43, 19.27, 19.17. HRMS (ESI+) Calcd for C15H27INaOSi (M + Na): 401.0774. Found: 401.0773.
Bis(2-(iodomethyl)pent-4-en-1-yl)diisopropoxysilane (2). To a solution of tetraallylsilane (0.19 g, 1.0 mmol) in DCM (10 mL), I2 (0.76 g, 3.0 mmol) was added, and the mixture was stirred for 6 h. The solution was then cooled to 0 °C before triethylamine (3.5 mmol) and isopropanol (0.21 mL, 2.5 mmol) were added, and the resulting mixture was stirred and allowed to slowly warm to room temperature over 6 h. The reaction was quenched with water (15 mL) and extracted with DCM (2 × 15 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated on a rotary evaporator. The crude product was purified with column chromatography on silica (20:1 to 10:1 Hexanes:EtOAc), giving 2 (0.42 g, 85%) as an oil and ~1:1 mixture of diastereomers. Spectral data for the mixture of two diastereomers: IR (ATR) 3072, 2975, 1638, 1589, 1428, 1380, 1219, 1109, 1021, 997, 915, 880 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.71 (ddtd, J = 17.1, 10.0, 7.2, 2.5 Hz, 4H), 5.13 (dq, J = 17.1, 1.7 Hz, 4H), 5.10–5.06 (m, 4H), 4.27–4.09 (m, 4H), 3.43–3.31 (m, 8H), 2.22 (dtdd, J = 12.5, 5.6, 2.6, 1.3 Hz, 4H), 2.11 (ddddd, J = 15.1, 7.4, 6.3, 2.5, 1.2 Hz, 4H), 1.59–1.51 (m, 4H), 1.29–1.15 (m, 28H), 0.68 (dd, J = 6.7, 1.4 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 137.91, 115.51, 64.77, 44.48, 28.60, 25.56, 22.33, 19.74. HRMS (ESI+) Calcd for C18H34I2NaO2Si (M + Na): 587.0315. Found: 587.0312.
(Cyclohexyloxy)bis(2-(iodomethyl)pent-4-en-1-yl)(isopropoxy)silane (3). To a solution of 1 (0.1 g, 0.26 mmol) in DCM (2.6 mL), iodine (0.078 g, 1.2 equiv.) was added, and the mixture was stirred for 6 h before cooling to 0 °C and triethylamine (0.09 mL, 2.5 equiv.) and cyclohexanol (0.04 mL, 1.5 equiv.) were added. The resulting mixture was allowed to slowly warm to room temperature before quenching with water (15 mL) and extracting with DCM (2 × 15 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated on a rotary evaporator. The crude product was purified with column chromatography on silica (20:1 to 10:1 Hexanes:EtOAc), giving 3 (0.13 g, 85%) as an oil and mixture of diastereomers. Spectral data for the mixture of three diastereomers: IR (ATR) 3068, 2972, 2951, 1610, 1569, 1418, 1410, 1353, 1311, 1201, 1109, 1021, 997, 914, 877, 732 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.88–5.77 (m, 2H), 5.72 (dqt, J = 17.2, 7.3, 2.6 Hz, 2H), 5.17–5.04 (m, 4H), 4.99–4.88 (m, 4H), 4.26–4.13 (m, 2H), 3.85–3.74 (m, 2H), 3.44–3.30 (m, 8H), 2.22 (dttd, J = 13.9, 5.6, 2.7, 1.4 Hz, 4H), 2.11 (dtdd, J = 13.9, 7.4, 2.6, 1.3 Hz, 4H), 1.81–1.77 (m, 4H), 1.75–1.69 (m, 4H), 1.63–1.47 (m, 8H), 1.40–1.24 (m, 10H), 1.21–1.16 (m, 14H), 0.71–0.66 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 13C NMR (126 MHz, CDCl3) δ 135.78, 135.67, 133.48, 117.29, 117.12, 114.39, 70.70, 65.08, 65.05, 65.03, 41.14, 41.12, 41.10, 41.07, 41.00, 40.96, 35.71, 34.75, 34.71, 34.57, 25.81, 25.72, 25.50, 23.92, 23.90, 22.41, 22.39, 19.79, 19.75, 19.70, 19.35, 19.33, 18.82, 18.80. HRMS (ESI+) Calcd for C21H38I2NaO2Si (M + Na): 627.0628. Found: 627.0631.
1-Allyl-3-(iodomethyl)-1-isopropoxy-2,3,4,7-tetrahydro-1H-silepine (4). To a solution of 1 (0.19 g, 0.5 mmol) in degassed DCM (25 mL), Ru-I (0.02 g, 0.025 mmol) was added, and the mixture was stirred for 15 h. The solution was then concentrated on a rotary evaporator, and the crude product was purified with chromatography on silica (20:1 to 10:1 Hexanes:MTBE) giving 4 (0.165 g, 94%) as an oil and ~1:1 mixture of diastereomers. Spectral data for the mixture of two diastereomers: IR (ATR) 3042, 2972, 2911, 1614, 1562, 1435, 1410, 1364, 1207, 1151, 1009, 998, 901, 876, 731 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.87–5.66 (m, 4H), 5.65–5.55 (m, 2H), 4.93 (dq, J = 6.3, 1.6 Hz, 1H), 4.91–4.87 (m, 3H), 4.03 (dq, J = 12.0, 6.0 Hz, 2H), 3.21 (dd, J = 5.5, 2.3 Hz, 1H), 3.19 (dd, J = 5.4, 2.2 Hz, 1H), 3.14 (dd, J = 9.5, 7.1 Hz, 2H), 2.29 (ddt, J = 14.2, 7.9, 2.1 Hz, 1H), 2.19 (ddt, J = 13.8, 8.1, 2.1 Hz, 1H), 2.09 (dtd, J = 13.8, 8.2, 1.4 Hz, 2H), 1.93 (ddtt, J = 12.6, 7.3, 3.3, 2.0 Hz, 1H), 1.88–1.75 (m, 2H), 1.72–1.63 (m, 3H), 1.62–1.53 (m, 4H), 1.23–1.17 (m, 2H), 1.16 (d, J = 5.9 Hz, 6H), 1.15 (d, J = 5.9 Hz, 6H), 0.76 (dd, J = 14.2, 11.3 Hz, 2H), 0.69 (dd, J = 14.6, 11.0 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 133.76, 133.56, 127.63, 126.88, 126.69, 126.52, 113.97, 113.87, 65.60, 65.36, 38.58, 36.58, 34.40, 34.21, 25.80, 25.78, 25.71, 24.34, 23.83, 23.48, 21.42, 20.06, 19.94, 14.87, 14.79. HRMS (ESI+) Calcd for C13H24IOSi (M + H) 351.0641. Found 351.0641.
Diallyl(allyloxy)(2-(iodomethyl)pent-4-en-1-yl)silane (5). To a solution of tetraallylsilane (0.2 g, 1.0 mmol) in DCM (10 mL), I2 (0.26 g, 1.0 mmol) was added, and the mixture was stirred for 6 h. The solution was then cooled to 0 °C before diisopropylethylamine (0.45 mL, 2.5 mmol) and allyl alcohol (0.11 mL, 1.5 mmol) were added, and the resulting mixture was stirred and allowed to slowly warm to room temperature over 6 h. The reaction was quenched with water (15 mL) and extracted with DCM (2 × 15 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated on a rotary evaporator. The crude product was purified with column chromatography on silica (20:1 to 10:1 Hexanes:EtOAc), giving 5 (0.29 g, 77%) as an oil. IR (ATR) 3076, 2972, 2909, 1629, 1418, 1219, 1157, 1031, 991, 826, 787 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.91 (ddt, J = 17.1, 10.4, 4.7 Hz, 1H), 5.82 (ddtd, J = 16.9, 10.1, 8.0, 5.3 Hz, 2H), 5.74–5.65 (m, 1H), 5.27 (dq, J = 17.1, 1.8 Hz, 1H), 5.17–5.05 (m, 3H), 5.00–4.91 (m, 4H), 4.23 (dq, J = 4.8, 1.7 Hz, 2H), 3.34 (dd, J = 9.7, 4.3 Hz, 1H), 3.29 (dd, J = 9.6, 5.2 Hz, 1H), 2.20 (dddt, J = 13.7, 6.9, 5.4, 1.4 Hz, 1H), 2.10 (dtt, J = 13.9, 7.4, 1.1 Hz, 1H), 1.80–1.66 (m, 4H), 1.60 (dddd, J = 11.6, 6.8, 5.3, 1.7 Hz, 1H), 0.77 (d, J = 6.8 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 136.66, 135.48, 133.13, 117.46, 114.74, 114.73, 114.61, 64.26, 41.16, 34.51, 21.82, 21.78, 19.20, 18.95. HRMS (ESI+) Calcd for C15H25INaOSi (M + Na) 399.0617. Found 399.0618.
11-(Iodomethyl)-1-oxa-6-silaspiro[5.6]dodeca-3,8-diene (6a). To a solution of 5 (0.05 g, 0.13 mmol) in degassed DCM (6.5 mL), Ru-I (0.008 g, 0.013 mmol) was added, and the mixture was stirred for 15 h. The solution was then concentrated on a rotary evaporator, and the crude product was purified with chromatography on silica (20:1 to 10:1 Hexanes:MTBE) giving 6a (0.031 g, 75%) as an oil and ~1:1 mixture of diastereomers. Spectral data for the mixture of two diastereomers: IR (ATR) 2955, 2930, 2875, 1460, 1352, 1344, 1254, 1143, 1103, 1004, 969, 835, 774, 741, 724, 666 cm−1. 1H NMR (500 MHz, CDCl3) δ 5.85 (dddd, J = 13.7, 7.5, 5.1, 2.8 Hz, 2H), 5.78 (dt, J = 10.6, 7.3 Hz, 2H), 5.72–5.62 (m, 2H), 5.61–5.56 (m, 2H), 4.41 (dp, J = 5.2, 2.7 Hz, 4H), 3.21 (ddd, J = 9.8, 5.4, 4.6 Hz, 2H), 3.15 (ddd, J = 9.7, 7.2, 2.8 Hz, 2H), 2.32 (ddt, J = 14.3, 7.7, 1.9 Hz, 1H), 2.24–2.08 (m, 4H), 2.06–1.97 (m, 1H), 1.95–1.87 (m, 1H), 1.81 (dddddd, J = 11.1, 9.2, 7.4, 5.6, 3.7, 1.9 Hz, 1H), 1.77–1.63 (m, 2H), 1.40–1.19 (m, 6H), 0.85 (dd, J = 14.2, 11.1 Hz, 1H), 0.79 (dd, J = 14.6, 10.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 128.74, 128.70, 128.29, 127.17, 126.40, 126.03, 123.71, 123.48, 63.32, 63.30, 39.11, 36.72, 34.27, 33.97, 25.08, 24.62, 19.38, 19.34, 15.54, 15.40, 11.22, 9.58. HRMS (ESI+) Calcd for C11H17INaOSi (M + Na) 342.9991. Found 342.9990.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25189996/s1.
Author Contributions
Conceptualization, G.W.O.; methodology, G.W.O., E.D.T. and K.E.W.; investigation, G.W.O., E.D.T. and K.E.W.; resources, G.W.O.; data curation, G.W.O., E.D.T. and K.E.W.; writing—original draft preparation, G.W.O.; writing—review and editing, E.D.T. and K.E.W.; supervision, G.W.O.; project administration, G.W.O.; funding acquisition, G.W.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the American Chemical Society Petroleum Research Fund (62228-UR1).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- O’Neil, G.W.; Cummins, E.J. Iodine-mediated rearrangements of diallylsilanes. Tetrahedron Lett. 2017, 58, 3406–3409. [Google Scholar] [PubMed]
- Hosomi, A.; Sakurai, H. Protection of alcohols and acids with allylsilanes catalyzed by iodine or iodotrimethylsilane in chlorinated hydrocarbons. Chem. Lett. 1981, 10, 85–88. [Google Scholar]
- Jones, R.G.; Cragg, R.H.; Swain, A.C. Structure and mechanism in the cyclopolymerization of diallylsilanes. Eur. Polym. J. 1992, 28, 651–655. [Google Scholar]
- Suslova, E.N.; Albanov, B.A.; Shainyan, J. Unusual product of the Si-C bond cleavage in diallyldiphenylsilane. Russ. J. Gen. Chem. 2008, 78, 1016–1017. [Google Scholar]
- Suslova, E.N.; Albanov, B.A.; Shainyan, J. Transformations of diallylsilanes under the action of electrophilic reagents. J. Organomet. Chem. 2009, 694, 420–426. [Google Scholar]
- Bols, M.; Skrydstrup, T. Silicon-Tethered Reactions. Chem. Rev. 1995, 95, 1253–1277. [Google Scholar]
- Bracegirdle, S.; Anderson, E.A. Recent advances in the use of temporary silicon tethers in metal-mediated reactions. Chem. Soc. Rev. 2010, 39, 4114–4129. [Google Scholar]
- Moss, G.P. ‘stereogenic unit’. In IUPAC Compendium of Chemical Terminology, 3rd ed.; International Union of Pure and Applied Chemistry: Research Triangle Park, NC, USA, 2006; Online version 3.0.1, 2019. [Google Scholar] [CrossRef]
- Xu, L.-W.; Li, L.; Lai, G.-Q.; Jiang, J.-X. The recent synthesis and application of silicon-stereogenic silanes: A renewed and significant challenge in asymmetric synthesis. Chem. Soc. Rev. 2011, 40, 1777–1790. [Google Scholar]
- Wu, Y.; Zheng, L.; Wang, Y.; Wang, P. Catalytic asymmetric synthesis of silicon-stereogenic organosilanes. Chem 2023, 9, 3461–3514. [Google Scholar]
- Ye, Z.-T.; Wu, Z.-W.; Zhang, X.-X.; Zhou, J.; Yu, J.-S. Organocatalytic enantioselective construction of Si-stereocenters: Recent advances and perspectives. Chem. Soc. Rev. 2024, 53, 8546–8562. [Google Scholar]
- Tomooka, K.; Nakazaki, A.; Nakai, T. A Novel Aryl Migration from Silicon to Carbon: An Efficient Approach to Asymmetric Synthesis of r-Aryl â-Hydroxy Cyclic Amines and Silanols. J. Am. Chem. Soc. 2000, 122, 408–409. [Google Scholar]
- Igawa, K.; Takada, J.; Shimono, T.; Tomooka, K. Enantioselective Synthesis of Silanol. J. Am. Chem. Soc. 2008, 130, 16132–16133. [Google Scholar] [PubMed]
- Bauer, J.O.; Strohmann, C. Stereocontrol in Nucleophilic Substitution Reactions at Silicon: The Role of Permutation in Generating Silicon-Centered Chirality. J. Am. Chem. Soc. 2015, 137, 4304–4307. [Google Scholar]
- Bai, X.-F.; Zou, J.-F.; Chen, M.-Y.; Xu, Z.; Li, L.; Cui, Y.-M.; Zheng, Z.-J.; Xu, L.-W. Lewis-Base-Mediated Diastereoselective Silylations of Alcohols: Synthesis of Silicon-Stereogenic Dialkoxysilanes Controlled by Chiral Aryl BINMOLs. Chem. Asian J. 2017, 12, 1730–1735. [Google Scholar]
- Zhang, X.-X.; Gao, Y.; Zhang, Y.-X.; Zhou, J.; Yu, J.-S. Highly Enantioselective Construction of Multifunctional Silicon- Stereogenic Silacycles by Asymmetric Enamine Catalysis. Angew. Chem. Int. Ed. 2023, e202217724. [Google Scholar]
- Bains, W.; Tacke, R. Silicon chemistry as a novel source of chemical diversity in drug design. Curr. Opin. Drug Discovery Dev. 2003, 6, 526–543. [Google Scholar]
- Fotie, J.; Matherne, C.M.; Wroblewski, J.E. Silicon switch: Carbon-silicon Bioisosteric replacement as a strategy to modulate the selectivity, physicochemical, and drug-like properties in anticancer pharmacophores. Chem. Biol. Drug. Des. 2023, 102, 235–254. [Google Scholar]
- Kawakami, Y.; Kakihana, Y.; Ooi, O.; Oishi, M.; Suzuki, K.; Shinke, S.; Uenishi, K. Control of stereochemical structures of silicon-containing polymeric systems. Polym. Int. 2009, 58, 279–284. [Google Scholar]
- Fomina, I.A.; Myers, C.R.; Soumis, C.L.M.; Scheuermann, M.L.; McCarty, J.; Clark, T.B.; O’Neil, G.W. Regiodivergent Medium-Ring Oxasilacycle Synthesis from Diallylsilanes. Heterocycles 2022, 104, 1966–1993. [Google Scholar] [CrossRef]
- Ahmad, I.; Falck-Pederson, M.L.; Undheim, K. Synthesis of silacycloalkenes and silaspirenes by Ru(II)-catalyzed ring-closing metathesis reactions. J. Organomet. Chem. 2001, 625, 160–172. [Google Scholar]
- Mahieux, C.; Laguerre, M.; Landais, Y.; Pianet, I. Multinuclear magnetic resonance and molecular modeling investigations as unambiguous methods for the determination of silacycle 3D structures. Magn. Reson. Chem. 2004, 42, 467–473. [Google Scholar] [PubMed]
- Anhaus, J.T.; Gibson, V.C.; Clegg, W.; Collingwood, S.P. Silicon-Containing Macrocycles and Polymers via Metathesis: X-ray Crystal Structures of cis,cis- and trans,trans-1,1,6,6,Tetraphenyl-1,6-disilacyclodeca-3,8-diene. Organometallics 1993, 12, 1780–1798. [Google Scholar]
- Kulkarni, A.A.; Diver, S.T. Cycloheptadiene Ring Synthesis by Tandem Intermolecular Enyne Metathesis. Org. Lett. 2003, 5, 3463–3466. [Google Scholar]
- Shieh, P.; Nguyen, H.V.-T.; Johnson, J.A. Tailored silyl ether monomers enable backbond-degradable polynorbornene-based linear, bottlebrush and star copolymers through ROMP. Nature Chem. 2019, 11, 1124–1132. [Google Scholar]
- Morontsev, A.; Gringolts, M.; Lakhtin, V.; Finkelshtein, E. Synthesis of high-molecular weight poly(1,1-dimethyl-1-silapentene) by olefin metathesis polymerization in the presence of Grubbs catalysts. J. Organomet. Chem. 2020, 911, 121156. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).