
Germline DNA Damage Repair Gene Alterations in Patients with Metachronous Breast and Colorectal Cancer

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
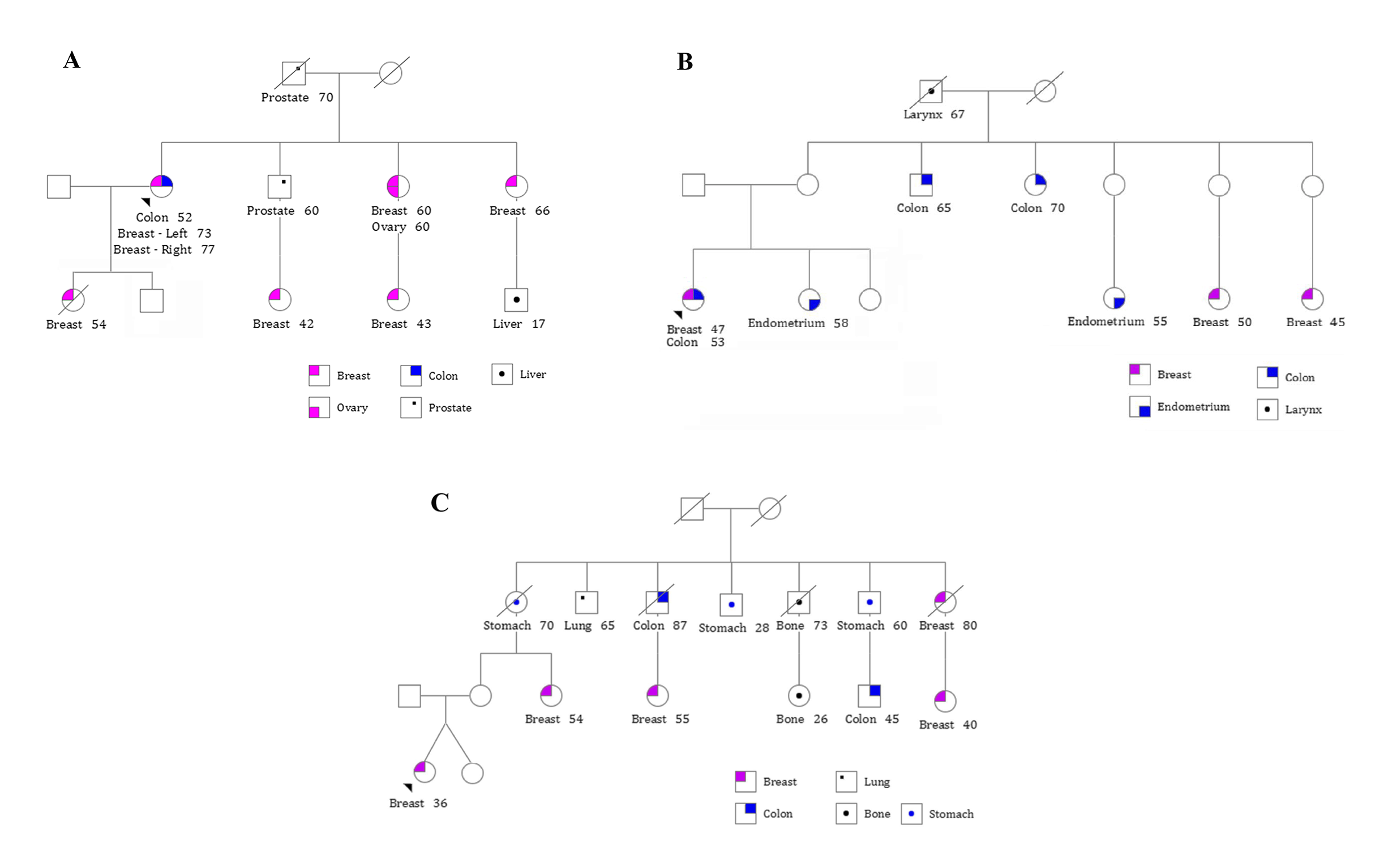
2.1. Clinical and Pathological Data of the Patients
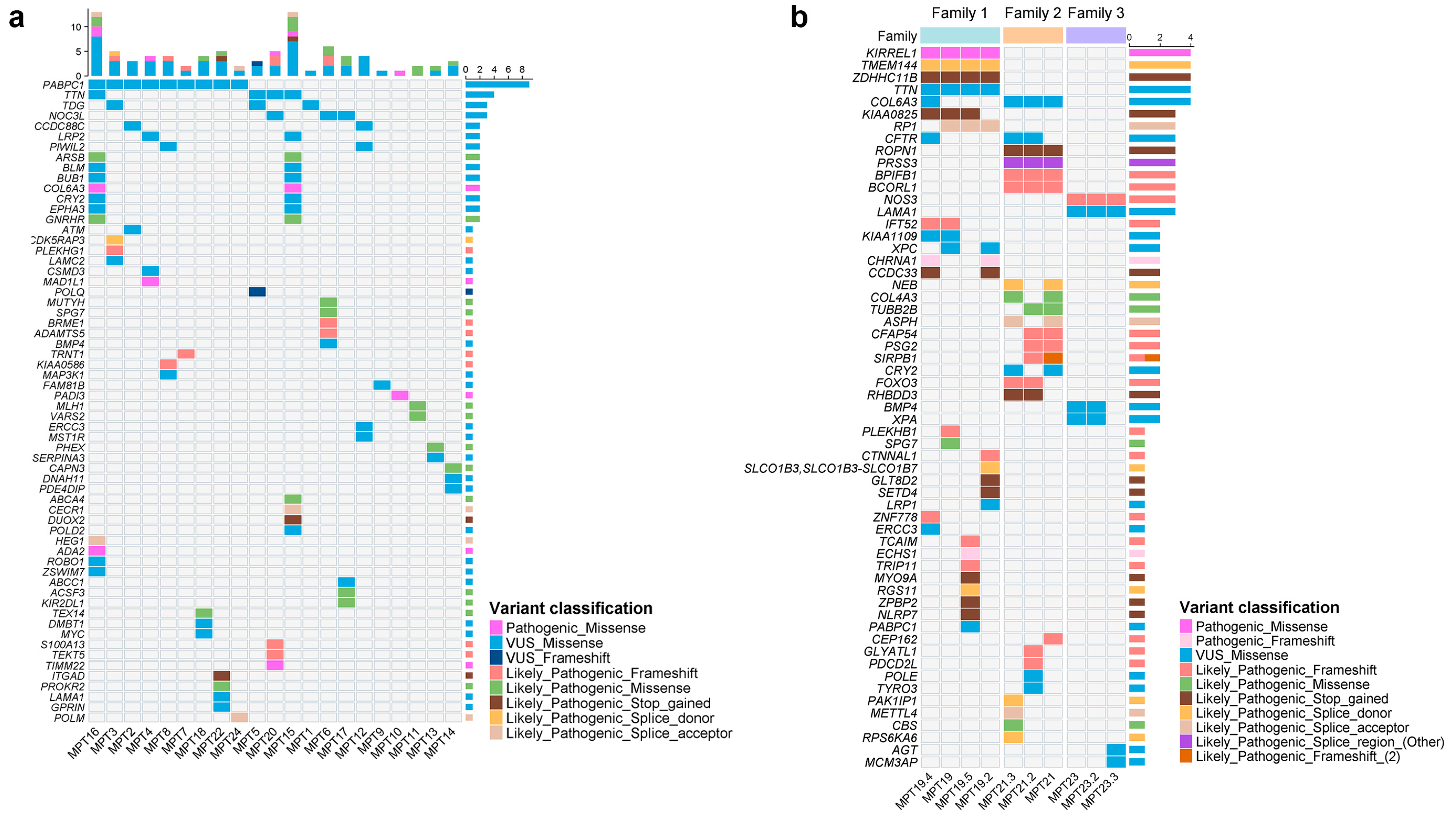
2.2. Single-Nucleotide Variants (SNVs) and InDels
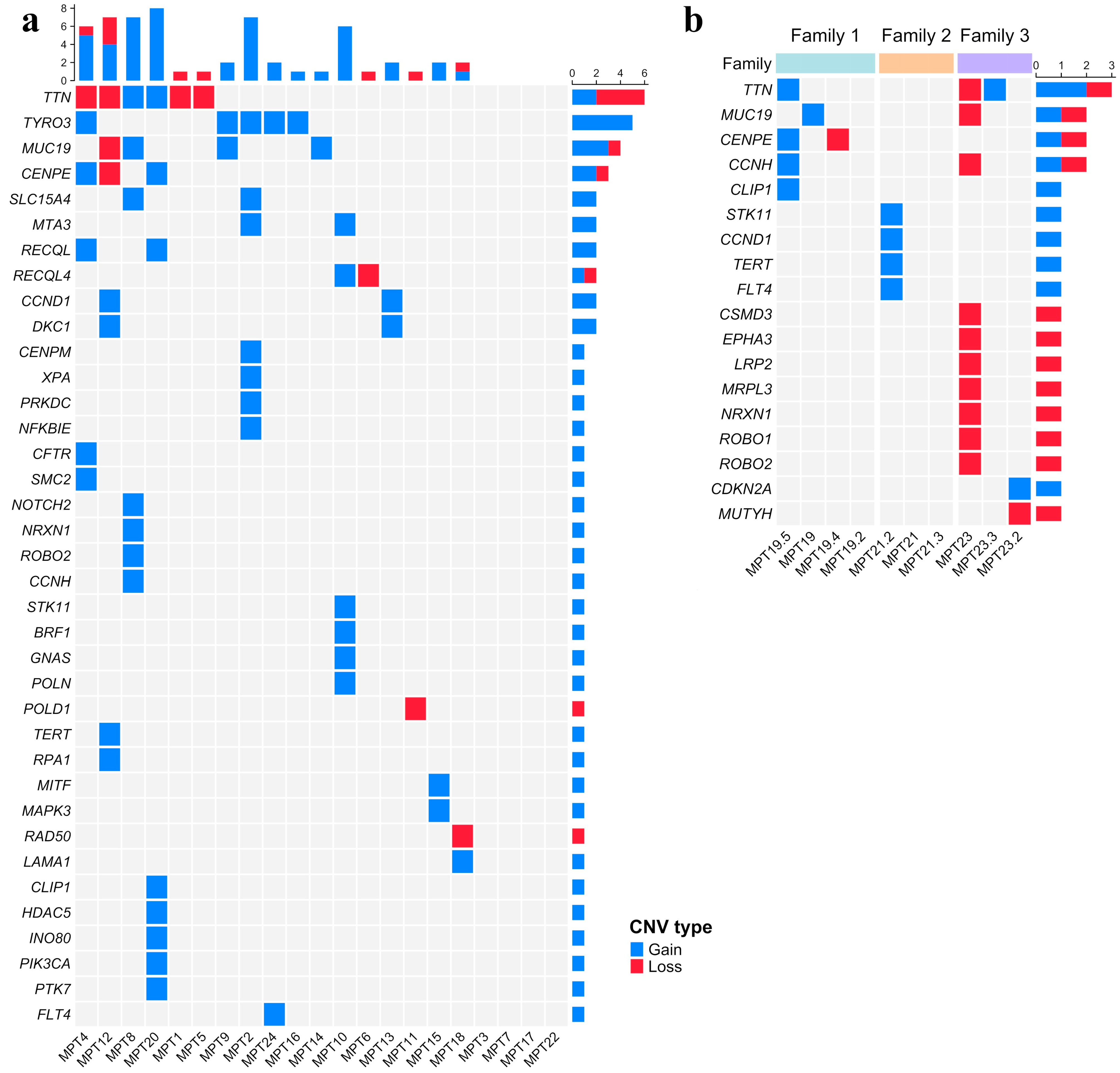
2.3. Copy Number Variations (CNVs)
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Isolation and Whole Exome Sequencing (WES)
4.3. Data Analysis and SNV and CNV Prioritization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A. Increasing Incidence of Early-Onset Colorectal Cancer. N. Engl. J. Med. 2022, 386, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Halamkova, J.; Kazda, T.; Pehalova, L.; Gonec, R.; Kozakova, S.; Bohovicova, L.; Krakorova, D.A.; Slaby, O.; Demlova, R.; Svoboda, M.; et al. Second primary malignancies in colorectal cancer patients. Sci. Rep. 2021, 11, 2759. [Google Scholar] [CrossRef] [PubMed]
- Molina-Montes, E.; Requena, M.; Sánchez-Cantalejo, E.; Fernández, M.F.; Arroyo-Morales, M.; Espín, J.; Arrebola, J.P.; Sánchez, M.J. Risk of second cancers cancer after a first primary breast cancer: A systematic review and meta-analysis. Gynecol. Oncol. 2015, 136, 158–171. [Google Scholar] [CrossRef]
- Goldgar, D.E.; Easton, D.F.; Cannon-Albright, L.A.; Skolnick, M.H. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J. Natl. Cancer Inst. 1994, 86, 1600–1608. [Google Scholar] [CrossRef]
- Dong, C.; Hemminki, K. Multiple primary cancers of the colon, breast and skin (melanoma) as models for polygenic cancers. Int. J. Cancer 2001, 92, 883–887. [Google Scholar] [CrossRef]
- Villacis, R.A.; Abreu, F.B.; Miranda, P.M.; Domingues, M.A.; Carraro, D.M.; Santos, E.M.; Andrade, V.P.; Rossi, B.M.; Achatz, M.I.; Rogatto, S.R. ROBO1 deletion as a novel germline alteration in breast and colorectal cancer patients. Tumour Biol. 2016, 37, 3145–3153. [Google Scholar] [CrossRef]
- Côrtes, L.; Basso, T.R.; Villacis, R.A.R.; Souza, J.D.S.; Jørgensen, M.M.A.; Achatz, M.I.; Rogatto, S.R. Co-Occurrence of Germline Genomic Variants and Copy Number Variations in Hereditary Breast and Colorectal Cancer Patients. Genes 2023, 14, 1580. [Google Scholar] [CrossRef]
- Copur, M.S.; Manapuram, S. Multiple Primary Tumors Over a Lifetime. Oncology 2019, 33, 629384. [Google Scholar]
- Lee, N.Y.; Lee, N.Y.; Hum, M.; Amali, A.A.; Lim, W.K.; Wong, M.; Myint, M.K.; Tay, R.J.; Ong, P.Y.; Samol, J.; et al. Whole-exome sequencing of BRCA-negative breast cancer patients and case-control analyses identify variants associated with breast cancer susceptibility. Hum. Genom. 2022, 16, 61. [Google Scholar] [CrossRef]
- Fatemi, N.; Tu, S.J.; Chung, C.C.; Moghadam, P.K.; Mojarad, E.N.; Sadeghi, A.; Totonchi, M.; Aghdaei, H.A.; Chang, J.G. Whole exome sequencing identifies MAP3K1, MSH2, and MLH1 as potential cancer-predisposing genes in familial early-onset colorectal cancer. Kaohsiung J. Med. Sci. 2023, 39, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Marks, J.H.; Mandell, J.B.; New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Angeli, D.; Salvi, S.; Tedaldi, G. Genetic Predisposition to Breast and Ovarian Cancers: How Many and Which Genes to Test? Int. J. Mol. Sci. 2020, 21, 1128. [Google Scholar] [CrossRef]
- Breast Cancer Association Consortium; Mavaddat, N.; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Keeman, R.; Bolla, M.K.; Dennis, J.; Wang, Q.; et al. Pathology of tumors associated with pathogenic germline variants in 9 breast cancer susceptibility genes. JAMA Oncol. 2022, 8, e216744. [Google Scholar] [CrossRef]
- Sopik, V.; Phelan, C.; Cybulski, C.; Narod, S.A. BRCA1 and BRCA2 mutations and the risk for colorectal cancer. Clin. Genet. 2015, 87, 411–418. [Google Scholar] [CrossRef]
- Cullinane, C.M.; Creavin, B.; O’Connell, E.P.; Kelly, L.; O’Sullivan, M.J.; Corrigan, M.A.; Redmond, H.P. Risk of colorectal cancer associated with BRCA1 and/or BRCA2 mutation carriers: Systematic review and meta-analysis. Br. J. Surg. 2020, 107, 951–959. [Google Scholar] [CrossRef]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Rebuzzi, F.; Ulivi, P.; Tedaldi, G. Genetic Predisposition to Colorectal Cancer: How Many and Which Genes to Test? Int. J. Mol. Sci. 2023, 24, 2137. [Google Scholar] [CrossRef]
- Win, A.K.; Lindor, N.M.; Jenkins, M.A. Risk of breast cancer in Lynch syndrome: A systematic review. Breast Cancer Res. 2013, 15, R27. [Google Scholar] [CrossRef]
- Sheehan, M.; Heald, B.; Yanda, C.; Kelly, E.D.; Grobmyer, S.; Eng, C.; Kalady, M.; Pederson, H. Investigating the Link between Lynch Syndrome and Breast Cancer. Eur. J. Breast Health 2020, 16, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.J.; da Silva, E.M.; Marra, A.; Gazzo, A.M.; Selenica, P.; Rai, V.K.; Mandelker, D.; Pareja, F.; Misyura, M.; D’Alfonso, T.M.; et al. Morphologic and Genomic Characteristics of Breast Cancers Occurring in Individuals with Lynch Syndrome. Clin. Cancer Res. 2022, 28, 404–413. [Google Scholar] [CrossRef]
- Felicio, P.S.; Grasel, R.S.; Campacci, N.; de Paula, A.E.; Galvão, H.C.R.; Torrezan, G.T.; Sabato, C.S.; Fernandes, G.C.; Souza, C.P.; Michelli, R.D.; et al. Whole-exome sequencing of non-BRCA1/BRCA2 mutation carrier cases at high-risk for hereditary breast/ovarian cancer. Hum. Mutat. 2021, 42, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Skopelitou, D.; Srivastava, A.; Miao, B.; Kumar, A.; Dymerska, D.; Paramasivam, N.; Schlesner, M.; Lubinski, J.; Hemminki, K.; Försti, A.; et al. Whole exome sequencing identifies novel germline variants of SLC15A4 gene as potentially cancer predisposing in familial colorectal cancer. Mol. Genet. Genom. 2022, 297, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Pande, M.; Joon, A.; Brewster, A.M.; Chen, W.V.; Hopper, J.L.; Eng, C.; Shete, S.; Casey, G.; Schumacher, F.; Lin, Y.; et al. Genetic susceptibility markers for a breast-colorectal cancer phenotype: Exploratory results from genome-wide association studies. PLoS ONE 2018, 13, e0196245. [Google Scholar] [CrossRef]
- Akcay, I.M.; Celik, E.; Agaoglu, N.B.; Alkurt, G.; Kizilboga Akgun, T.; Yildiz, J.; Enc, F.; Kir, G.; Canbek, S.; Kilic, A.; et al. Germline pathogenic variant spectrum in 25 cancer susceptibility genes in Turkish breast and colorectal cancer patients and elderly controls. Int. J. Cancer 2021, 148, 285–295. [Google Scholar] [CrossRef]
- Infante, M.; Arranz-Ledo, M.; Lastra, E.; Olaverri, A.; Ferreira, R.; Orozco, M.; Hernández, L.; Martínez, N.; Durán, M. Profiling of the genetic features of patients with breast, ovarian, colorectal and extracolonic cancers: Association to CHEK2 and PALB2 germline mutations. Clin. Chim. Acta 2024, 552, 117695. [Google Scholar] [CrossRef]
- NCCN Guidelines—Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 3. 2024. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 15 March 2024).
- NCCN Guidelines—Genetic/Familial High-Risk Assessment: Colorectal Version 2. 2023. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf (accessed on 15 March 2024).
- Meijers-Heijboer, H.; Wijnen, J.; Vasen, H.; Wasielewski, M.; Wagner, A.; Hollestelle, A.; Elstrodt, F.; van den Bos, R.; de Snoo, A.; Fat, G.T.; et al. The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am. J. Hum. Genet. 2003, 72, 1308–1314. [Google Scholar] [CrossRef]
- Naseem, H.; Boylan, J.; Speake, D.; Leask, K.; Shenton, A.; Lalloo, F.; Hill, J.; Trump, D.; Evans, D.G. Inherited association of breast and colorectal cancer: Limited role of CHEK2 compared with high-penetrance genes. Clin. Genet. 2006, 70, 388–395. [Google Scholar] [CrossRef]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef]
- Tudini, E.; Davidson, A.L.; Dressel, U.; Andrews, L.; Antill, Y.; Crook, A.; Field, M.; Gattas, M.; Harris, R.; Kirk, J.; et al. Implementing gene curation for hereditary cancer susceptibility in Australia: Achieving consensus on genes with clinical utility. J. Med. Genet. 2021, 58, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Lee, K.; Abul-Husn, N.S.; Amendola, L.M.; Brothers, K.; Chung, W.K.; Gollob, M.H.; Gordon, A.S.; Harrison, S.M.; Hershberger, R.E.; et al. ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2023, 25, 100866. [Google Scholar] [CrossRef] [PubMed]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the Expanding Landscape of Clinical Actionability for Patients with Cancer. Cancer Discov. 2024, 14, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Broderick, P.; Bagratuni, T.; Vijayakrishnan, J.; Lubbe, S.; Chandler, I.; Houlston, R.S. Evaluation of NTHL1, NEIL1, NEIL2, MPG, TDG, UNG and SMUG1 genes in familial colorectal cancer predisposition. BMC Cancer 2006, 6, 243. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Rozadilla, C.; Álvarez-Barona, M.; Quintana, I.; López-Novo, A.; Amigo, J.; Cameselle-Teijeiro, J.M.; Roman, E.; Gonzalez, D.; Llor, X.; Bujanda, L.; et al. Exome sequencing of early-onset patients supports genetic heterogeneity in colorectal cancer. Sci. Rep. 2021, 11, 11135. [Google Scholar] [CrossRef]
- Tedaldi, G.; Tebaldi, M.; Zampiga, V.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Cangini, I.; Pirini, F.; Petracci, E.; Rocca, A.; et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 2017, 8, 47064–47075. [Google Scholar] [CrossRef]
- Bonache, S.; Esteban, I.; Moles-Fernández, A.; Tenés, A.; Duran-Lozano, L.; Montalban, G.; Bach, V.; Carrasco, E.; Gadea, N.; López-Fernández, A.; et al. Multigene panel testing beyond BRCA1/2 in breast/ovarian cancer Spanish families and clinical actionability of findings. J. Cancer Res. Clin. Oncol. 2018, 144, 2495–2513. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, Q.; Yin, X.; Zhu, X.; Zhao, L.; Zhang, Z.; Wei, R.; Wang, B.; Li, X. Association of XPA polymorphism with breast cancer risk: A meta-analysis. Medicine 2018, 97, e11276. [Google Scholar] [CrossRef]
- Malik, S.S.; Zia, A.; Rashid, S.; Mubarik, S.; Masood, N.; Hussain, M.; Yasmin, A.; Bano, R. XPC as breast cancer susceptibility gene: Evidence from genetic profiling, statistical inferences and protein structural analysis. Breast Cancer 2020, 27, 1168–1176. [Google Scholar] [CrossRef]
- Shahi, R.B.; De Brakeleer, S.; Caljon, B.; Pauwels, I.; Bonduelle, M.; Joris, S.; Fontaine, C.; Vanhoeij, M.; Van Dooren, S.; Teugels, E.; et al. Identification of candidate cancer predisposing variants by performing whole-exome sequencing on index patients from BRCA1 and BRCA2-negative breast cancer families. BMC Cancer 2019, 19, 313. [Google Scholar] [CrossRef]
- Chan, A.B.; Parico, G.C.G.; Fribourgh, J.L.; Ibrahim, L.H.; Bollong, M.J.; Partch, C.L.; Lamia, K.A. CRY2 missense mutations suppress P53 and enhance cell growth. Proc. Natl. Acad. Sci. USA 2021, 118, e2101416118. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline Pathogenic Variants in the Ataxia telangiectasia Mutated (ATM) Gene Are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev. Res. 2021, 14, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, C.M.; do Canto, L.M.; Villacis, R.A.R.; Petersen, A.H.; Aagaard, M.M.; Cury, S.S.; Formiga, M.N.D.C.; Junior, S.A.; Rogatto, S.R. The repertoire of germline variants in patients with early-onset rectal cancer. Cancer Commun. 2022, 42, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.R.; Doyle, M.A.; Ryland, G.L.; Rowley, S.M.; Choong, D.Y.; Tothill, R.W.; Thorne, H.; kConFab; Barnes, D.R.; Li, J.; et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012, 8, e1002894. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Gay, M.; Franch-Expósito, S.; Arnau-Collell, C.; Park, S.; Supek, F.; Muñoz, J.; Bonjoch, L.; Gratacós-Mulleras, A.; Sánchez-Rojas, P.A.; Esteban-Jurado, C.; et al. Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer. Cancers 2019, 11, 362. [Google Scholar] [CrossRef]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Spier, I.; Kerick, M.; Drichel, D.; Horpaopan, S.; Altmüller, J.; Laner, A.; Holzapfel, S.; Peters, S.; Adam, R.; Zhao, B.; et al. Exome sequencing identifies potential novel candidate genes in patients with unexplained colorectal adenomatous polyposis. Fam. Cancer 2016, 15, 281–288. [Google Scholar] [CrossRef]
- Soares de Lima, Y.; Arnau-Collell, C.; Díaz-Gay, M.; Bonjoch, L.; Franch-Expósito, S.; Muñoz, J.; Moreira, L.; Ocaña, T.; Cuatrecasas, M.; Herrera-Pariente, C.; et al. Germline and Somatic Whole-Exome Sequencing Identifies New Candidate Genes Involved in Familial Predisposition to Serrated Polyposis Syndrome. Cancers 2021, 13, 929. [Google Scholar] [CrossRef]
- Raskin, L.; Guo, Y.; Du, L.; Clendenning, M.; Rosty, C.; Colon Cancer Family Registry (CCFR); Lindor, N.M.; Gruber, S.B.; Buchanan, D.D. Targeted sequencing of established and candidate colorectal cancer genes in the Colon Cancer Family Registry Cohort. Oncotarget 2017, 8, 93450–93463. [Google Scholar] [CrossRef]
- Dos Santos, W.; de Andrade, E.S.; Garcia, F.A.O.; Campacci, N.; Sábato, C.D.S.; Melendez, M.E.; Reis, R.M.; Galvão, H.C.R.; Palmero, E.I. Whole-Exome Sequencing Identifies Pathogenic Germline Variants in Patients with Lynch-Like Syndrome. Cancers 2022, 14, 4233. [Google Scholar] [CrossRef]
- Matejcic, M.; Shaban, H.A.; Quintana, M.W.; Schumacher, F.R.; Edlund, C.K.; Naghi, L.; Pai, R.K.; Haile, R.W.; Levine, A.J.; Buchanan, D.D.; et al. Rare Variants in the DNA Repair Pathway and the Risk of Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2021, 30, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wang, Y.; Tang, Y.; Chen, Z.; Hou, J.; Dai, J.; Wang, Y.; Wang, L.; Xu, H.; Tian, B.; et al. Mechanism of genome instability mediated by human DNA polymerase mu misincorporation. Nat. Commun. 2021, 12, 3759. [Google Scholar] [CrossRef]
- Paluch-Shimon, S.; Evron, E. Targeting DNA repair in breast cancer. Breast 2019, 47, 33–42. [Google Scholar] [CrossRef]
- Gentile, D.; Losurdo, A.; Sagona, A.; Zuradelli, M.; Gatzemeier, W.; Barbieri, E.; Testori, A.; Errico, V.; Bianchi, P.; Biondi, E.; et al. Surgical management of BRCA-mutation carriers: A single institution experience. Eur. J. Surg. Oncol. 2022, 48, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Guan, J.; Li, G.M. DNA mismatch repair in cancer immunotherapy. NAR Cancer 2023, 5, zcad031. [Google Scholar] [CrossRef] [PubMed]
- Minguillón, J.; Ramírez, M.J.; Rovirosa, L.; Bustamante-Madrid, P.; Camps-Fajol, C.; Ruiz de Garibay, G.; Shimelis, H.; Montanuy, H.; Pujol, R.; Hernandez, G.; et al. CDK5RAP3, a New BRCA2 Partner That Regulates DNA Repair, Is Associated with Breast Cancer Survival. Cancers 2022, 14, 353. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, X.; Liu, T.; Liu, X.; Geng, J.; He, X.; Liu, Y.; Pang, D. Increased expression of mitotic arrest deficient-like 1 (MAD1L1) is associated with poor prognosis and insensitive to Taxol treatment in breast cancer. Breast Cancer Res. Treat. 2013, 140, 323–330. [Google Scholar] [CrossRef]
- Singh, A.K.; Talseth-Palmer, B.; Xavier, A.; Scott, R.J.; Drabløs, F.; Sjursen, W. Detection of germline variants with pathogenic potential in 48 patients with familial colorectal cancer by using whole exome sequencing. BMC Med. Genom. 2023, 16, 126. [Google Scholar] [CrossRef]
- Sawazaki, R.; Imai, S.; Yokogawa, M.; Hosoda, N.; Hoshino, S.I.; Mio, M.; Mio, K.; Shimada, I.; Osawa, M. Characterization of the multimeric structure of poly(A)-binding protein on a poly(A) tail. Sci. Rep. 2018, 8, 1455. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Li, X.; Lou, H.; Geng, T.; Jin, T.; Liang, P.; Li, S.; Long, Y.; Chen, C. Genetic association of PLCE1, C11orf92-C11orf93, and NOC3L with colorectal cancer risk in the Han population. Tumour Biol. 2014, 35, 1813–1817. [Google Scholar] [CrossRef] [PubMed]
- Marouf, C.; Göhler, S.; Filho, M.I.; Hajji, O.; Hemminki, K.; Nadifi, S.; Försti, A. Analysis of functional germline variants in APOBEC3 and driver genes on breast cancer risk in Moroccan study population. BMC Cancer 2016, 16, 165. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Moya, A.; Morales, S.; Arancibia, T.; Gonzalez-Hormazabal, P.; Tapia, J.C.; Godoy-Herrera, R.; Reyes, J.M.; Gomez, F.; Waugh, E.; Jara, L. Germline Variants in Driver Genes of Breast Cancer and Their Association with Familial and Early-Onset Breast Cancer Risk in a Chilean Population. Cancers 2020, 12, 249. [Google Scholar] [CrossRef]
- Wei, H.; Ren, K.; Zhang, Q.; Jin, Y.; Cao, B.; Tian, Z.; Mao, T.; Ren, L. Titin as a potential novel therapeutic target in colorectal cancer. J. Cell Mol. Med. 2023, 27, 2937–2944. [Google Scholar] [CrossRef]
- Kumar, R.; Nagpal, G.; Kumar, V.; Usmani, S.S.; Agrawal, P.; Raghava, G.P.S. HumCFS: A database of fragile sites in human chromosomes. BMC Genom. 2019, 19, 985. [Google Scholar] [CrossRef]
- Simpson, B.S.; Pye, H.; Whitaker, H.C. The oncological relevance of fragile sites in cancer. Commun. Biol. 2021, 4, 567. [Google Scholar] [CrossRef]
- Juhari, W.K.W.; Ahmad Amin Noordin, K.B.; Zakaria, A.D.; Rahman, W.F.W.A.; Mokhter, W.M.M.W.M.; Hassan, M.R.A.; Sidek, A.S.M.; Zilfalil, B.A. Whole-Genome Profiles of Malay Colorectal Cancer Patients with Intact MMR Proteins. Genes 2021, 12, 1448. [Google Scholar] [CrossRef]
- DeRycke, M.S.; Gunawardena, S.R.; Middha, S.; Asmann, Y.W.; Schaid, D.J.; McDonnell, S.K.; Riska, S.M.; Eckloff, B.W.; Cunningham, J.M.; Fridley, B.L.; et al. Identification of novel variants in colorectal cancer families by high-throughput exome sequencing. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1239–1251. [Google Scholar] [CrossRef]
- BenAyed-Guerfali, D.; Kifagi, C.; BenKridis-Rejeb, W.; Ammous-Boukhris, N.; Ayedi, W.; Khanfir, A.; Daoud, J.; Mokdad-Gargouri, R. The Identification by Exome Sequencing of Candidate Genes in BRCA-Negative Tunisian Patients at a High Risk of Hereditary Breast/Ovarian Cancer. Genes 2022, 13, 1296. [Google Scholar] [CrossRef]
- Koivuluoma, S.; Tervasmäki, A.; Kauppila, S.; Winqvist, R.; Kumpula, T.; Kuismin, O.; Moilanen, J.; Pylkäs, K. Exome sequencing identifies a recurrent variant in SERPINA3 associating with hereditary susceptibility to breast cancer. Eur. J. Cancer 2021, 143, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Radmanesh, H.; Liu, D.; Geffers, R.; Shandiz, F.H.; Sadr-Nabavi, A.; Hillemanns, P.; Park-Simon, T.W.; Dörk, T. Exome sequencing identifies RASSF1 and KLK3 germline variants in an Iranian multiple-case breast cancer family. Eur. J. Med. Genet. 2022, 65, 104425. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Jermusyk, A.; Wu, L.; Hoskins, J.W.; Collins, I.; Mocci, E.; Zhang, M.; Song, L.; Chung, C.C.; Zhang, T.; et al. A Transcriptome-Wide Association Study Identifies Novel Candidate Susceptibility Genes for Pancreatic Cancer. J. Natl. Cancer Inst. 2020, 112, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.J.; Jeon, Y.J.; Kim, J.W.; Chong, S.Y.; Hong, S.P.; Oh, D.; Cho, Y.K.; Chung, K.W.; Kim, N.K. Association of eNOS polymorphisms (-786T>C, 4a4b, 894G>T) with colorectal cancer susceptibility in the Korean population. Gene 2013, 512, 275–281. [Google Scholar] [CrossRef]
- Gao, X.; Wang, J.; Wang, W.; Wang, M.; Zhang, J. eNOS Genetic Polymorphisms and Cancer Risk: A Meta-Analysis and a Case-Control Study of Breast Cancer. Medicine 2015, 94, e972. [Google Scholar] [CrossRef]
- Banno, K.; Kisu, I.; Yanokura, M.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Hirasawa, A.; Aoki, D. Hereditary gynecological tumors associated with Peutz-Jeghers syndrome (Review). Oncol. Lett. 2013, 6, 1184–1188. [Google Scholar] [CrossRef]
- Mohammadi, L.; Vreeswijk, M.P.; Oldenburg, R.; van den Ouweland, A.; Oosterwijk, J.C.; van der Hout, A.H.; Hoogerbrugge, N.; Ligtenberg, M.; Ausems, M.G.; van der Luijt, R.B.; et al. A simple method for co-segregation analysis to evaluate the pathogenicity of unclassified variants; BRCA1 and BRCA2 as an example. BMC Cancer 2009, 9, 211. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar]
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Sondka, Z.; Dhir, N.B.; Carvalho-Silva, D.; Jupe, S.; Madhumita; McLaren, K.; Starkey, M.; Ward, S.; Wilding, J.; Ahmed, M.; et al. COSMIC: A curated database of somatic variants and clinical data for cancer. Nucleic Acids Res. 2024, 52, D1210–D1217. [Google Scholar] [CrossRef]
- Griffith, M.; Spies, N.C.; Krysiak, K.; McMichael, J.F.; Coffman, A.C.; Danos, A.M.; Ainscough, B.J.; Ramirez, C.A.; Rieke, D.T.; Kujan, L.; et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 2017, 49, 170–174. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| DNA Repair Pathway | Gene a | Type of Alteration | Case |
|---|---|---|---|
| Base excision repair (BER) | MUTYH | SNV (LP) | MPT6 |
| TDG | SNV (VUS) | MPT1, MPT3, and MPT5 | |
| Nucleotide excision repair (NER) | ERCC3 | SNV (VUS) | MPT12 |
| XPC | SNV (VUS) | MPT19 | |
| XPA | SNV (VUS)/CNV | MPT2 and MPT23 | |
| CRY2 | SNV (VUS) | MPT15, MPT16, and MPT21 | |
| CCNH | CNV | MPT8 and MPT23 | |
| Mismatch repair (MMR) | MLH1 | SNV (LP/VUS) | MPT11 |
| Homologous recombination (HR) | ATM | SNV (VUS) | MPT2 |
| BLM | SNV (VUS) | MPT15 and MPT16 | |
| ZSWIM7 | SNV (VUS) | MPT16 | |
| POLQ | SNV (VUS) | MPT5 | |
| POLD1 a | CNV | MPT11 | |
| POLD2 a | SNV (VUS) | MPT15 | |
| RAD50 | CNV | MPT18 | |
| RECQL b | CNV | MPT4 and MPT20 | |
| RECQL4 | CNV | MPT6 and MPT10 | |
| POLN | CNV | MPT10 | |
| INO80 | CNV | MPT20 | |
| RPA1 c | CNV | MPT12 | |
| Non-homologous end-joining repair (NHEJ) | POLM | SNV (LP) | MPT24 |
| PRKDC | CNV | MPT2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villacis, R.A.R.; Côrtes, L.; Basso, T.R.; do Canto, L.M.; Souza, J.S.; Aagaard, M.M.; da Cruz Formiga, M.N.; Aguiar, S., Jr.; Achatz, M.I.; Rogatto, S.R. Germline DNA Damage Repair Gene Alterations in Patients with Metachronous Breast and Colorectal Cancer. Int. J. Mol. Sci. 2024, 25, 10275. https://doi.org/10.3390/ijms251910275
Villacis RAR, Côrtes L, Basso TR, do Canto LM, Souza JS, Aagaard MM, da Cruz Formiga MN, Aguiar S Jr., Achatz MI, Rogatto SR. Germline DNA Damage Repair Gene Alterations in Patients with Metachronous Breast and Colorectal Cancer. International Journal of Molecular Sciences. 2024; 25(19):10275. https://doi.org/10.3390/ijms251910275
Chicago/Turabian StyleVillacis, Rolando André Rios, Luiza Côrtes, Tatiane Ramos Basso, Luisa Matos do Canto, Jeferson Santos Souza, Mads Malik Aagaard, Maria Nirvana da Cruz Formiga, Samuel Aguiar, Jr., Maria Isabel Achatz, and Silvia Regina Rogatto. 2024. "Germline DNA Damage Repair Gene Alterations in Patients with Metachronous Breast and Colorectal Cancer" International Journal of Molecular Sciences 25, no. 19: 10275. https://doi.org/10.3390/ijms251910275
APA StyleVillacis, R. A. R., Côrtes, L., Basso, T. R., do Canto, L. M., Souza, J. S., Aagaard, M. M., da Cruz Formiga, M. N., Aguiar, S., Jr., Achatz, M. I., & Rogatto, S. R. (2024). Germline DNA Damage Repair Gene Alterations in Patients with Metachronous Breast and Colorectal Cancer. International Journal of Molecular Sciences, 25(19), 10275. https://doi.org/10.3390/ijms251910275