
Navigating the Controversies: Role of TRPM Channels in Pain States



Abstract
:
1. Introduction
2. Transient Receptor Potential (TRP) Channels
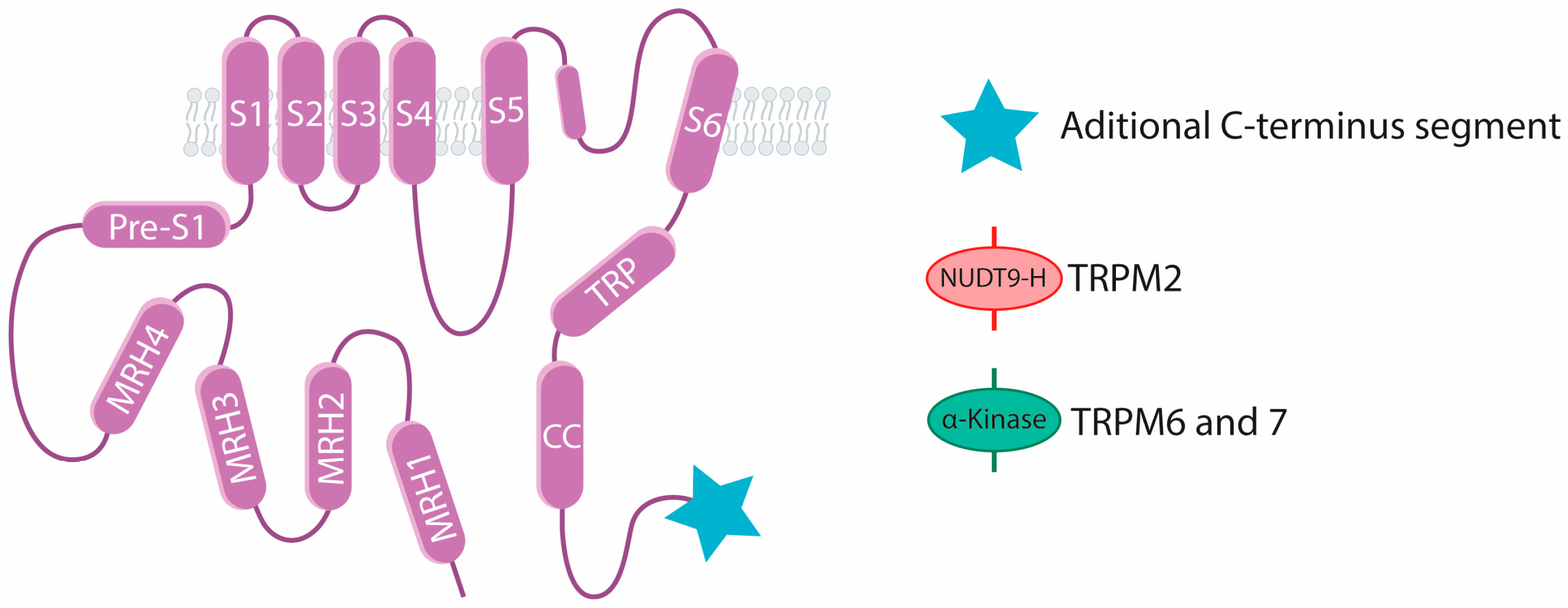
3. The TRPM Channels Subfamily
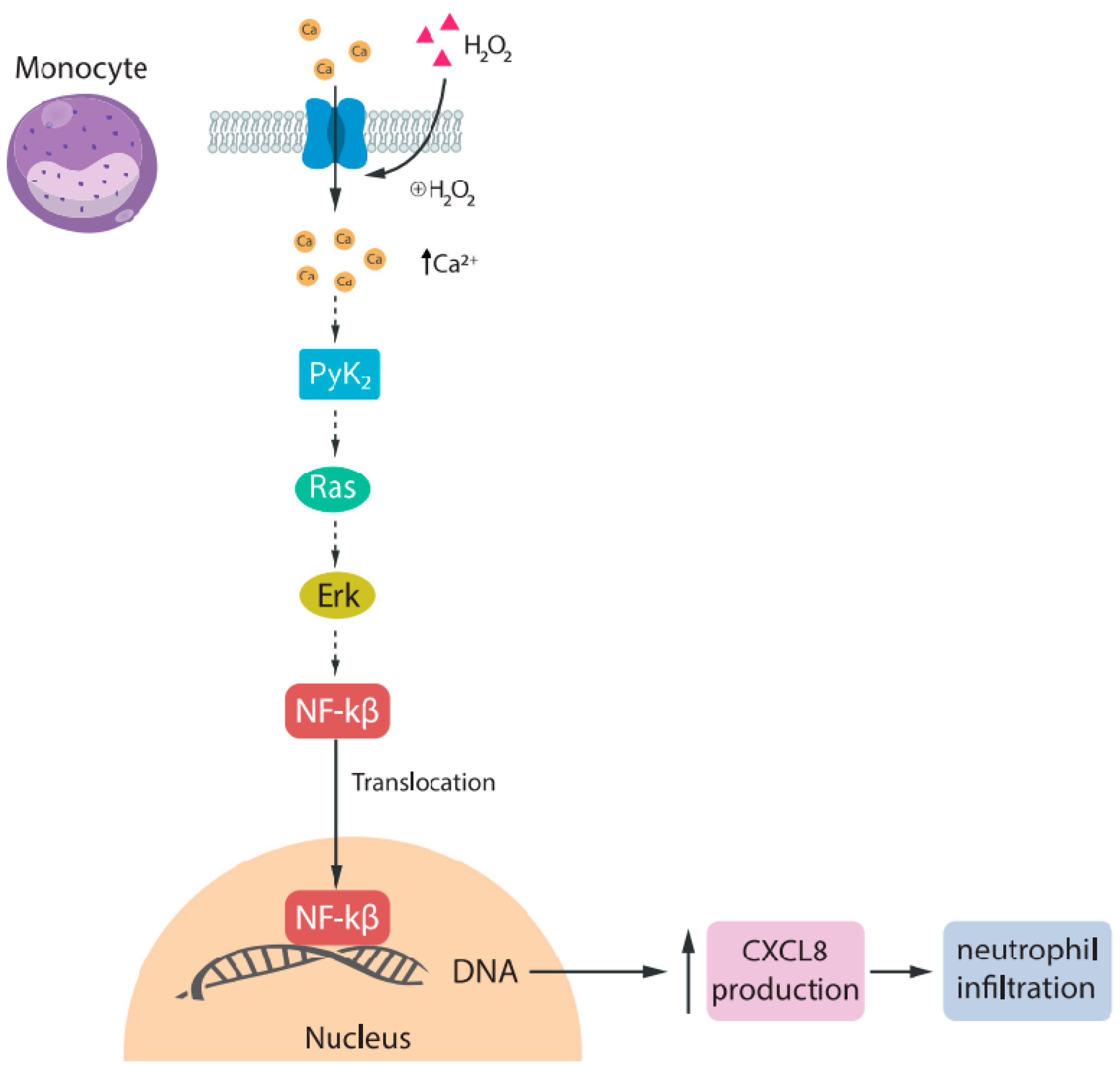
3.1. TRPM2
3.2. TRPM3
3.3. TRPM8
4. TRPM Channels in Cancer Pain
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bingham, B.; Ajit, S.K.; Blake, D.R.; Samad, T.A. The molecular basis of pain and its clinical implications in rheumatology. Nat. Clin. Pract. Rheumatol. 2009, 5, 28–37. [Google Scholar] [CrossRef]
- Kuner, R. Central mechanisms of pathological pain. Nat. Med. 2010, 16, 1258–1266. [Google Scholar] [CrossRef]
- Ellison, D.L. Physiology of Pain. Crit. Care Nurs. Clin. N. Am. 2017, 29, 397–406. [Google Scholar] [CrossRef]
- Bourinet, E.; Altier, C.; Hildebrand, M.E.; Trang, T.; Salter, M.W.; Zamponi, G.W. Calcium-permeable ion channels in pain signaling. Physiol. Rev. 2014, 94, 81–140. [Google Scholar] [CrossRef]
- Bhuiyan, S.A.; Xu, M.; Yang, L.; Semizoglou, E.; Bhatia, P.; Pantaleo, K.I.; Tochitsky, I.; Jain, A.; Erdogan, B.; Blair, S.; et al. Harmonized cross-species cell atlases of trigeminal and dorsal root ganglia. Sci. Adv. 2024, 10, eadj9173. [Google Scholar] [CrossRef]
- Liu, J.A.; Yu, J.; Cheung, C.W. Immune Actions on the Peripheral Nervous System in Pain. Int. J. Mol. Sci. 2021, 22, 1448. [Google Scholar] [CrossRef]
- Baral, P.; Udit, S.; Chiu, I.M. Pain and immunity: Implications for host defence. Nat. Rev. Immunol. 2019, 19, 433–447. [Google Scholar] [CrossRef]
- Gao, Y.J.; Ji, R.R. Targeting astrocyte signaling for chronic pain. Neurotherapeutics 2010, 7, 482–493. [Google Scholar] [CrossRef]
- Zhang, M.; Ma, Y.; Ye, X.; Zhang, N.; Pan, L.; Wang, B. TRP (transient receptor potential) ion channel family: Structures, biological functions and therapeutic interventions for diseases. Signal Transduct. Target. Ther. 2023, 8, 261. [Google Scholar]
- Yan, Q.; Gao, C.; Li, M.; Lan, R.; Wei, S.; Fan, R.; Cheng, W. TRP Ion Channels in Immune Cells and Their Implications for Inflammation. Int. J. Mol. Sci. 2024, 25, 2719. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef]
- Himmel, N.J.; Cox, D.N. Transient receptor potential channels: Current perspectives on evolution, structure, function and nomenclature. Proc. Biol. Sci. 2020, 287, 20201309. [Google Scholar] [CrossRef]
- Montell, C.; Birnbaumer, L.; Flockerzi, V.; Bindels, R.J.; Bruford, E.A.; Caterina, M.J.; Clapham, D.E.; Harteneck, C.; Heller, S.; Julius, D.; et al. A unified nomenclature for the superfamily of TRP cation channels. Mol. Cell 2002, 9, 229–231. [Google Scholar] [CrossRef]
- Rosenbaum, T.; Morales-Lazaro, S.L.; Islas, L.D. TRP channels: A journey towards a molecular understanding of pain. Nat. Rev. Neurosci. 2022, 23, 596–610. [Google Scholar] [CrossRef]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lu, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85, 102111. [Google Scholar] [CrossRef]
- Chubanov, V.; Kottgen, M.; Touyz, R.M.; Gudermann, T. TRPM channels in health and disease. Nat. Rev. Nephrol. 2024, 20, 175–187. [Google Scholar] [CrossRef]
- Zhang, Z.; Toth, B.; Szollosi, A.; Chen, J.; Csanady, L. Structure of a TRPM2 channel in complex with Ca(2+) explains unique gating regulation. eLife 2018, 7, e36409. [Google Scholar] [CrossRef]
- Yin, Y.; Wu, M.; Hsu, A.L.; Borschel, W.F.; Borgnia, M.J.; Lander, G.C.; Lee, S.Y. Visualizing structural transitions of ligand-dependent gating of the TRPM2 channel. Nat. Commun. 2019, 10, 3740. [Google Scholar] [CrossRef]
- Huang, Y.; Winkler, P.A.; Sun, W.; Lu, W.; Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 2018, 562, 145–149. [Google Scholar] [CrossRef]
- Iordanov, I.; Toth, B.; Szollosi, A.; Csanady, L. Enzyme activity and selectivity filter stability of ancient TRPM2 channels were simultaneously lost in early vertebrates. eLife 2019, 8, e44556. [Google Scholar] [CrossRef]
- Wang, L.; Fu, T.M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362, eaav4809. [Google Scholar] [CrossRef]
- Yu, X.; Xie, Y.; Zhang, X.; Ma, C.; Liu, L.; Zhen, W.; Xu, L.; Zhang, J.; Liang, Y.; Zhao, L.; et al. Structural and functional basis of the selectivity filter as a gate in human TRPM2 channel. Cell Rep. 2021, 37, 110025. [Google Scholar] [CrossRef]
- Zhao, C.; MacKinnon, R. Structural and functional analyses of a GPCR-inhibited ion channel TRPM3. Neuron 2023, 111, 81–91.e7. [Google Scholar] [CrossRef]
- Winkler, P.A.; Huang, Y.; Sun, W.; Du, J.; Lu, W. Electron cryo-microscopy structure of a human TRPM4 channel. Nature 2017, 552, 200–204. [Google Scholar] [CrossRef]
- Guo, J.; She, J.; Zeng, W.; Chen, Q.; Bai, X.C.; Jiang, Y. Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 2017, 552, 205–209. [Google Scholar] [CrossRef]
- Autzen, H.E.; Myasnikov, A.G.; Campbell, M.G.; Asarnow, D.; Julius, D.; Cheng, Y. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 2018, 359, 228–232. [Google Scholar] [CrossRef]
- Duan, J.; Li, Z.; Li, J.; Santa-Cruz, A.; Sanchez-Martinez, S.; Zhang, J.; Clapham, D.E. Structure of full-length human TRPM4. Proc. Natl. Acad. Sci. USA 2018, 115, 2377–2382. [Google Scholar] [CrossRef]
- Ruan, Z.; Haley, E.; Orozco, I.J.; Sabat, M.; Myers, R.; Roth, R.; Du, J.; Lu, W. Structures of the TRPM5 channel elucidate mechanisms of activation and inhibition. Nat. Struct. Mol. Biol. 2021, 28, 604–613. [Google Scholar] [CrossRef]
- Duan, J.; Li, Z.; Li, J.; Hulse, R.E.; Santa-Cruz, A.; Valinsky, W.C.; Abiria, S.A.; Krapivinsky, G.; Zhang, J.; Clapham, D.E. Structure of the mammalian TRPM7, a magnesium channel required during embryonic development. Proc. Natl. Acad. Sci. USA 2018, 115, E8201–E8210. [Google Scholar] [CrossRef]
- Nadezhdin, K.D.; Correia, L.; Narangoda, C.; Patel, D.S.; Neuberger, A.; Gudermann, T.; Kurnikova, M.G.; Chubanov, V.; Sobolevsky, A.I. Structural mechanisms of TRPM7 activation and inhibition. Nat. Commun. 2023, 14, 2639. [Google Scholar] [CrossRef]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science 2018, 359, 237–241. [Google Scholar] [CrossRef]
- Diver, M.M.; Cheng, Y.; Julius, D. Structural insights into TRPM8 inhibition and desensitization. Science 2019, 365, 1434–1440. [Google Scholar] [CrossRef]
- Zhao, C.; Xie, Y.; Xu, L.; Ye, F.; Xu, X.; Yang, W.; Yang, F.; Guo, J. Structures of a mammalian TRPM8 in closed state. Nat. Commun. 2022, 13, 3113. [Google Scholar] [CrossRef]
- Fruhwald, J.; Camacho Londono, J.; Dembla, S.; Mannebach, S.; Lis, A.; Drews, A.; Wissenbach, U.; Oberwinkler, J.; Philipp, S.E. Alternative splicing of a protein domain indispensable for function of transient receptor potential melastatin 3 (TRPM3) ion channels. J. Biol. Chem. 2012, 287, 36663–36672. [Google Scholar] [CrossRef]
- Held, K.; Aloi, V.D.; Freitas, A.C.N.; Janssens, A.; Segal, A.; Przibilla, J.; Philipp, S.E.; Wang, Y.T.; Voets, T.; Vriens, J. Pharmacological properties of TRPM3 isoforms are determined by the length of the pore loop. Br. J. Pharmacol. 2022, 179, 3560–3575. [Google Scholar] [CrossRef]
- Behrendt, M. TRPM3 in the eye and in the nervous system—From new findings to novel mechanisms. Biol. Chem. 2022, 403, 859–868. [Google Scholar] [CrossRef]
- Rohacs, T. Phosphoinositide Regulation of TRP Channels: A Functional Overview in the Structural Era. Annu. Rev. Physiol. 2024, 86, 329–355. [Google Scholar] [CrossRef]
- Hasan, R.; Zhang, X. Ca(2+) Regulation of TRP Ion Channels. Int. J. Mol. Sci. 2018, 19, 1256. [Google Scholar] [CrossRef] [PubMed]
- Talavera, K.; Yasumatsu, K.; Voets, T.; Droogmans, G.; Shigemura, N.; Ninomiya, Y.; Margolskee, R.F.; Nilius, B. Heat activation of TRPM5 underlies thermal sensitivity of sweet taste. Nature 2005, 438, 1022–1025. [Google Scholar] [CrossRef]
- Dhaka, A.; Murray, A.N.; Mathur, J.; Earley, T.J.; Petrus, M.J.; Patapoutian, A. TRPM8 is required for cold sensation in mice. Neuron 2007, 54, 371–378. [Google Scholar] [CrossRef]
- Tan, C.H.; McNaughton, P.A. TRPM2 and warmth sensation. Pflug. Arch. 2018, 470, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Vandewauw, I.; De Clercq, K.; Mulier, M.; Held, K.; Pinto, S.; Van Ranst, N.; Segal, A.; Voet, T.; Vennekens, R.; Zimmermann, K.; et al. A TRP channel trio mediates acute noxious heat sensing. Nature 2018, 555, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Csanady, L.; Torocsik, B. Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but intracellularly of the gate. J. Gen. Physiol. 2009, 133, 189–203. [Google Scholar] [CrossRef]
- Sumoza-Toledo, A.; Penner, R. TRPM2: A multifunctional ion channel for calcium signalling. J. Physiol. 2011, 589 Pt 7, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Kozai, D.; Kobayashi, R.; Ebert, M.; Mori, Y. Roles of TRPM2 in oxidative stress. Cell Calcium 2011, 50, 279–287. [Google Scholar] [CrossRef]
- Haraguchi, K.; Kawamoto, A.; Isami, K.; Maeda, S.; Kusano, A.; Asakura, K.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; Kaneko, S. TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J. Neurosci. 2012, 32, 3931–3941. [Google Scholar] [CrossRef]
- Rittner, H.L.; Mousa, S.A.; Labuz, D.; Beschmann, K.; Schafer, M.; Stein, C.; Brack, A. Selective local PMN recruitment by CXCL1 or CXCL2/3 injection does not cause inflammatory pain. J. Leukoc. Biol. 2006, 79, 1022–1032. [Google Scholar] [CrossRef]
- Lavich, T.R.; Siqueira Rde, A.; Farias-Filho, F.A.; Cordeiro, R.S.; Rodrigues e Silva, P.M.; Martins, M.A. Neutrophil infiltration is implicated in the sustained thermal hyperalgesic response evoked by allergen provocation in actively sensitized rats. Pain 2006, 125, 180–187. [Google Scholar] [CrossRef]
- Naziroglu, M. TRPM2 cation channels, oxidative stress and neurological diseases: Where are we now? Neurochem. Res. 2011, 36, 355–366. [Google Scholar] [CrossRef]
- Schafers, M.; Sorkin, L.S.; Geis, C.; Shubayev, V.I. Spinal nerve ligation induces transient upregulation of tumor necrosis factor receptors 1 and 2 in injured and adjacent uninjured dorsal root ganglia in the rat. Neurosci. Lett. 2003, 347, 179–182. [Google Scholar] [CrossRef]
- Andersson, D.A.; Gentry, C.; Moss, S.; Bevan, S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J. Neurosci. 2008, 28, 2485–2494. [Google Scholar] [CrossRef] [PubMed]
- Keeble, J.E.; Bodkin, J.V.; Liang, L.; Wodarski, R.; Davies, M.; Fernandes, E.S.; Coelho Cde, F.; Russell, F.; Graepel, R.; Muscara, M.N.; et al. Hydrogen peroxide is a novel mediator of inflammatory hyperalgesia, acting via transient receptor potential vanilloid 1-dependent and independent mechanisms. Pain 2009, 141, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Han, X.B.; Liu, X.; Hsueh, W.; De Plaen, I.G. Macrophage inflammatory protein-2 mediates the bowel injury induced by platelet-activating factor. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1220–G1226. [Google Scholar] [CrossRef] [PubMed]
- Wehrhahn, J.; Kraft, R.; Harteneck, C.; Hauschildt, S. Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J. Immunol. 2010, 184, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Kiya, T.; Gong, H.; Gao, X.; Malik, A.B. Role of the phagosomal redox-sensitive TRP channel TRPM2 in regulating bactericidal activity of macrophages. J. Cell Sci. 2017, 130, 735–744. [Google Scholar] [CrossRef]
- Di, A.; Gao, X.P.; Qian, F.; Kawamura, T.; Han, J.; Hecquet, C.; Ye, R.D.; Vogel, S.M.; Malik, A.B. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 2011, 13, 29–34. [Google Scholar] [CrossRef]
- Wang, G.; Cao, L.; Liu, X.; Sieracki, N.A.; Di, A.; Wen, X.; Chen, Y.; Taylor, S.; Huang, X.; Tiruppathi, C.; et al. Oxidant Sensing by TRPM2 Inhibits Neutrophil Migration and Mitigates Inflammation. Dev. Cell 2016, 38, 453–462. [Google Scholar] [CrossRef]
- Dorward, D.A.; Lucas, C.D.; Chapman, G.B.; Haslett, C.; Dhaliwal, K.; Rossi, A.G. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am. J. Pathol. 2015, 185, 1172–1184. [Google Scholar] [CrossRef]
- Oberwinkler, J.; Lis, A.; Giehl, K.M.; Flockerzi, V.; Philipp, S.E. Alternative splicing switches the divalent cation selectivity of TRPM3 channels. J. Biol. Chem. 2005, 280, 22540–22548. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Vangeel, L.; Benoit, M.; Miron, Y.; Miller, P.E.; De Clercq, K.; Chaltin, P.; Verfaillie, C.; Vriens, J.; Voets, T. Functional expression and pharmacological modulation of TRPM3 in human sensory neurons. Br. J. Pharmacol. 2020, 177, 2683–2695. [Google Scholar] [CrossRef]
- Vriens, J.; Held, K.; Janssens, A.; Toth, B.I.; Kerselaers, S.; Nilius, B.; Vennekens, R.; Voets, T. Opening of an alternative ion permeation pathway in a nociceptor TRP channel. Nat. Chem. Biol. 2014, 10, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Kichko, T.; De Clercq, K.; Klaassen, H.; Van Bree, R.; Vanherck, J.C.; Marchand, A.; Reeh, P.W.; Chaltin, P.; Voets, T.; et al. Activation of TRPM3 by a potent synthetic ligand reveals a role in peptide release. Proc. Natl. Acad. Sci. USA 2015, 112, E1363–E1372. [Google Scholar] [CrossRef] [PubMed]
- Oberwinkler, J. TRPM3, a biophysical enigma? Biochem. Soc. Trans. 2007, 35 Pt 1, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Oberwinkler, J.; Philipp, S.E. Trpm3. Handb. Exp. Pharmacol. 2014, 222, 427–459. [Google Scholar] [PubMed]
- Held, K.; Toth, B.I. TRPM3 in Brain (Patho)Physiology. Front. Cell Dev. Biol. 2021, 9, 635659. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.F.; Drews, A.; Loch, S.; Mohr, F.; Philipp, S.E.; Lambert, S.; Oberwinkler, J. TRPM3 channels provide a regulated influx pathway for zinc in pancreatic beta cells. Pflugers Arch. 2010, 460, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Badheka, D.; Yudin, Y.; Borbiro, I.; Hartle, C.M.; Yazici, A.; Mirshahi, T.; Rohacs, T. Inhibition of Transient Receptor Potential Melastatin 3 ion channels by G-protein betagamma subunits. eLife 2017, 6, e26147. [Google Scholar] [CrossRef]
- Su, S.; Yudin, Y.; Kim, N.; Tao, Y.X.; Rohacs, T. TRPM3 Channels Play Roles in Heat Hypersensitivity and Spontaneous Pain after Nerve Injury. J. Neurosci. 2021, 41, 2457–2474. [Google Scholar] [CrossRef]
- Krugel, U.; Straub, I.; Beckmann, H.; Schaefer, M. Primidone inhibits TRPM3 and attenuates thermal nociception in vivo. Pain 2017, 158, 856–867. [Google Scholar] [CrossRef]
- Alkhatib, O.; da Costa, R.; Gentry, C.; Quallo, T.; Bevan, S.; Andersson, D.A. Promiscuous G-Protein-Coupled Receptor Inhibition of Transient Receptor Potential Melastatin 3 Ion Channels by Gbetagamma Subunits. J. Neurosci. 2019, 39, 7840–7852. [Google Scholar] [CrossRef] [PubMed]
- Straub, I.; Krugel, U.; Mohr, F.; Teichert, J.; Rizun, O.; Konrad, M.; Oberwinkler, J.; Schaefer, M. Flavanones that selectively inhibit TRPM3 attenuate thermal nociception in vivo. Mol. Pharmacol. 2013, 84, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, W.; Qian, X.; Fang, Y.; Zhu, N. Liquiritigenin alleviates mechanical and cold hyperalgesia in a rat neuropathic pain model. Sci. Rep. 2014, 4, 5676. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Zhang, Y.; Yu, J. Antinociceptive Effects of Isosakuranetin in a Rat Model of Peripheral Neuropathy. Pharmacology 2017, 100, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Chen, J.; Sun, L.; Wu, S.; Gray, K.R.; Rich, A.; Huang, M.; Lin, J.H.; Feder, J.N.; Janovitz, E.B.; et al. Expression and characterization of human transient receptor potential melastatin 3 (hTRPM3). J. Biol. Chem. 2003, 278, 20890–20897. [Google Scholar] [CrossRef]
- Haring, M.; Zeisel, A.; Hochgerner, H.; Rinwa, P.; Jakobsson, J.E.T.; Lonnerberg, P.; La Manno, G.; Sharma, N.; Borgius, L.; Kiehn, O.; et al. Neuronal atlas of the dorsal horn defines its architecture and links sensory input to transcriptional cell types. Nat. Neurosci. 2018, 21, 869–880. [Google Scholar] [CrossRef]
- Sathyamurthy, A.; Johnson, K.R.; Matson, K.J.E.; Dobrott, C.I.; Li, L.; Ryba, A.R.; Bergman, T.B.; Kelly, M.C.; Kelley, M.W.; Levine, A.J. Massively Parallel Single Nucleus Transcriptional Profiling Defines Spinal Cord Neurons and Their Activity during Behavior. Cell Rep. 2018, 22, 2216–2225. [Google Scholar] [CrossRef]
- Wei, H.; Chen, Z.; Koivisto, A.; Pertovaara, A. Spinal mechanisms contributing to the development of pain hypersensitivity induced by sphingolipids in the rat. Pharmacol. Rep. 2021, 73, 672–679. [Google Scholar] [CrossRef]
- Shiels, A. TRPM3_miR-204: A complex locus for eye development and disease. Hum. Genom. 2020, 14, 7. [Google Scholar] [CrossRef]
- Aloi, V.D.; Pinto, S.; Van Bree, R.; Luyten, K.; Voets, T.; Vriens, J. TRPM3 as a novel target to alleviate acute oxaliplatin-induced peripheral neuropathic pain. Pain 2023, 164, 2060–2069. [Google Scholar] [CrossRef]
- Mulier, M.; Van Ranst, N.; Corthout, N.; Munck, S.; Vanden Berghe, P.; Vriens, J.; Voets, T.; Moilanen, L. Upregulation of TRPM3 in nociceptors innervating inflamed tissue. eLife 2020, 9, e61103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, Q.; Lian, Y.; Chen, Y. Botulinum toxin type A reduces the expression of transient receptor potential melastatin 3 and transient receptor potential vanilloid type 4 in the trigeminal subnucleus caudalis of a rat model of trigeminal neuralgia. Neuroreport 2019, 30, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Morenilla-Palao, C.; Planells-Cases, R.; Garcia-Sanz, N.; Ferrer-Montiel, A. Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J. Biol. Chem. 2004, 279, 25665–25672. [Google Scholar] [CrossRef]
- Dolly, J.O.; Aoki, K.R. The structure and mode of action of different botulinum toxins. Eur. J. Neurol. 2006, 13 (Suppl. S4), 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Shibata, M.; Toriumi, H.; Iwashita, T.; Funakubo, M.; Sato, H.; Kuroi, T.; Ebine, T.; Koizumi, K.; Suzuki, N. Reduction of TRPV1 expression in the trigeminal system by botulinum neurotoxin type-A. Neurobiol. Dis. 2012, 48, 367–378. [Google Scholar] [CrossRef]
- Dembla, S.; Behrendt, M.; Mohr, F.; Goecke, C.; Sondermann, J.; Schneider, F.M.; Schmidt, M.; Stab, J.; Enzeroth, R.; Leitner, M.G.; et al. Anti-nociceptive action of peripheral mu-opioid receptors by G-beta-gamma protein-mediated inhibition of TRPM3 channels. eLife 2017, 6, e26280. [Google Scholar] [CrossRef] [PubMed]
- Quallo, T.; Alkhatib, O.; Gentry, C.; Andersson, D.A.; Bevan, S. G protein betagamma subunits inhibit TRPM3 ion channels in sensory neurons. eLife 2017, 6, e26138. [Google Scholar] [CrossRef]
- Behrendt, M.; Gruss, F.; Enzeroth, R.; Dembla, S.; Zhao, S.; Crassous, P.A.; Mohr, F.; Nys, M.; Louros, N.; Gallardo, R.; et al. The structural basis for an on-off switch controlling Gbetagamma-mediated inhibition of TRPM3 channels. Proc. Natl. Acad. Sci. USA 2020, 117, 29090–29100. [Google Scholar] [CrossRef]
- Xie, M.X.; Cao, X.Y.; Zeng, W.A.; Lai, R.C.; Guo, L.; Wang, J.C.; Xiao, Y.B.; Zhang, X.; Chen, D.; Liu, X.G.; et al. ATF4 selectively regulates heat nociception and contributes to kinesin-mediated TRPM3 trafficking. Nat. Commun. 2021, 12, 1401. [Google Scholar] [CrossRef]
- Alonso-Carbajo, L.; Alpizar, Y.A.; Startek, J.B.; Lopez-Lopez, J.R.; Perez-Garcia, M.T.; Talavera, K. Activation of the cation channel TRPM3 in perivascular nerves induces vasodilation of resistance arteries. J. Mol. Cell Cardiol. 2019, 129, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, M.; Chen, Z.; Xu, Y.; Guo, L.; Wang, S.; Li, Y.; Shi, B.; Zhang, X.; Jin, X.D. TRPM3 channel activation inhibits contraction of the isolated human ureter via CGRP released from sensory nerves. Life Sci. 2021, 268, 118967. [Google Scholar] [CrossRef] [PubMed]
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Peier, A.M.; Moqrich, A.; Hergarden, A.C.; Reeve, A.J.; Andersson, D.A.; Story, G.M.; Earley, T.J.; Dragoni, I.; McIntyre, P.; Bevan, S.; et al. A TRP channel that senses cold stimuli and menthol. Cell 2002, 108, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.M.; Jordt, S.E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef]
- Colburn, R.W.; Lubin, M.L.; Stone, D.J., Jr.; Wang, Y.; Lawrence, D.; D’Andrea, M.R.; Brandt, M.R.; Liu, Y.; Flores, C.M.; Qin, N. Attenuated cold sensitivity in TRPM8 null mice. Neuron 2007, 54, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Thut, P.D.; Wrigley, D.; Gold, M.S. Cold transduction in rat trigeminal ganglia neurons in vitro. Neuroscience 2003, 119, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.T.; Seid, D.A. AG-3-5: A chemical producing sensations of cold. J. Pharm. Pharmacol. 1983, 35, 110–112. [Google Scholar] [CrossRef]
- Ding, Z.; Gomez, T.; Werkheiser, J.L.; Cowan, A.; Rawls, S.M. Icilin induces a hyperthermia in rats that is dependent on nitric oxide production and NMDA receptor activation. Eur. J. Pharmacol. 2008, 578, 201–208. [Google Scholar] [CrossRef]
- Rawls, S.M.; Gomez, T.; Ding, Z.; Raffa, R.B. Differential behavioral effect of the TRPM8/TRPA1 channel agonist icilin (AG-3-5). Eur. J. Pharmacol. 2007, 575, 103–104. [Google Scholar] [CrossRef]
- Xing, H.; Chen, M.; Ling, J.; Tan, W.; Gu, J.G. TRPM8 mechanism of cold allodynia after chronic nerve injury. J. Neurosci. 2007, 27, 13680–13690. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, C.J.; Garry, E.M.; Cottrell, D.F.; Rosie, R.; Anderson, H.; Robertson, D.C.; Fleetwood-Walker, S.M.; Mitchell, R. Analgesia mediated by the TRPM8 cold receptor in chronic neuropathic pain. Curr. Biol. 2006, 16, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Caspani, O.; Zurborg, S.; Labuz, D.; Heppenstall, P.A. The contribution of TRPM8 and TRPA1 channels to cold allodynia and neuropathic pain. PLoS ONE 2009, 4, e7383. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Zhou, S.; Kochukov, M.Y.; Westlund, K.N.; Carlton, S.M. Plasticity in intact A delta- and C-fibers contributes to cold hypersensitivity in neuropathic rats. Neuroscience 2007, 150, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Djouhri, L.; Wrigley, D.; Thut, P.D.; Gold, M.S. Spinal nerve injury increases the percentage of cold-responsive DRG neurons. Neuroreport 2004, 15, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Gauchan, P.; Andoh, T.; Kato, A.; Kuraishi, Y. Involvement of increased expression of transient receptor potential melastatin 8 in oxaliplatin-induced cold allodynia in mice. Neurosci. Lett. 2009, 458, 93–95. [Google Scholar] [CrossRef]
- Knowlton, W.M.; Palkar, R.; Lippoldt, E.K.; McCoy, D.D.; Baluch, F.; Chen, J.; McKemy, D.D. A sensory-labeled line for cold: TRPM8-expressing sensory neurons define the cellular basis for cold, cold pain, and cooling-mediated analgesia. J. Neurosci. 2013, 33, 2837–2848. [Google Scholar] [CrossRef]
- Kawashiri, T.; Egashira, N.; Kurobe, K.; Tsutsumi, K.; Yamashita, Y.; Ushio, S.; Yano, T.; Oishi, R. L type Ca(2)+ channel blockers prevent oxaliplatin-induced cold hyperalgesia and TRPM8 overexpression in rats. Mol. Pain 2012, 8, 7. [Google Scholar] [CrossRef]
- Chukyo, A.; Chiba, T.; Kambe, T.; Yamamoto, K.; Kawakami, K.; Taguchi, K.; Abe, K. Oxaliplatin-induced changes in expression of transient receptor potential channels in the dorsal root ganglion as a neuropathic mechanism for cold hypersensitivity. Neuropeptides 2018, 67, 95–101. [Google Scholar] [CrossRef]
- Descoeur, J.; Pereira, V.; Pizzoccaro, A.; Francois, A.; Ling, B.; Maffre, V.; Couette, B.; Busserolles, J.; Courteix, C.; Noel, J.; et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 2011, 3, 266–278. [Google Scholar] [CrossRef]
- Villalba-Riquelme, E.; de la Torre-Martinez, R.; Fernandez-Carvajal, A.; Ferrer-Montiel, A. Paclitaxel in vitro reversibly sensitizes the excitability of IB4(-) and IB4(+) sensory neurons from male and female rats. Br. J. Pharmacol. 2022, 179, 3693–3710. [Google Scholar] [CrossRef]
- Zhang, X.; Mak, S.; Li, L.; Parra, A.; Denlinger, B.; Belmonte, C.; McNaughton, P.A. Direct inhibition of the cold-activated TRPM8 ion channel by Galphaq. Nat. Cell Biol. 2012, 14, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Direct Galpha(q) Gating Is the Sole Mechanism for TRPM8 Inhibition Caused by Bradykinin Receptor Activation. Cell Rep. 2019, 27, 3672–3683.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yudin, Y.; Nagwekar, J.; Kang, C.; Shirokova, N.; Rohacs, T. G(alphaq) Sensitizes TRPM8 to Inhibition by PI(4,5)P(2) Depletion upon Receptor Activation. J. Neurosci. 2019, 39, 6067–6080. [Google Scholar] [CrossRef]
- Liu, L.; Rohacs, T. Regulation of the cold-sensing TRPM8 channels by phosphoinositides and G(q)-coupled receptors. Channels 2020, 14, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363, eaav9334. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, G.; Gkika, D.; Devilliers, M.; Kondratskyi, A.; Gordienko, D.; Busserolles, J.; Bokhobza, A.; Eschalier, A.; Skryma, R.; Prevarskaya, N. Opiates modulate thermosensation by internalizing cold receptor TRPM8. Cell Rep. 2013, 4, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Jasmin, L. Sustained Morphine Administration Induces TRPM8-Dependent Cold Hyperalgesia. J. Pain 2017, 18, 212–221. [Google Scholar] [CrossRef]
- Iftinca, M.; Basso, L.; Flynn, R.; Kwok, C.; Roland, C.; Hassan, A.; Defaye, M.; Ramachandran, R.; Trang, T.; Altier, C. Chronic morphine regulates TRPM8 channels via MOR-PKCbeta signaling. Mol. Brain 2020, 13, 61. [Google Scholar] [CrossRef]
- Liu, B.; Fan, L.; Balakrishna, S.; Sui, A.; Morris, J.B.; Jordt, S.E. TRPM8 is the principal mediator of menthol-induced analgesia of acute and inflammatory pain. Pain 2013, 154, 2169–2177. [Google Scholar] [CrossRef]
- Marini, M.; Titiz, M.; Souza Monteiro de Araujo, D.; Geppetti, P.; Nassini, R.; De Logu, F. TRP Channels in Cancer: Signaling Mechanisms and Translational Approaches. Biomolecules 2023, 13, 1557. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Banham, A.H.; Yaacob, N.S.; Nur Husna, S.M. The oncogenic roles of TRPM ion channels in cancer. J. Cell Physiol. 2019, 234, 14556–14573. [Google Scholar] [CrossRef] [PubMed]
- Middelbeek, J.; Kuipers, A.J.; Henneman, L.; Visser, D.; Eidhof, I.; van Horssen, R.; Wieringa, B.; Canisius, S.V.; Zwart, W.; Wessels, L.F.; et al. TRPM7 is required for breast tumor cell metastasis. Cancer Res. 2012, 72, 4250–4261. [Google Scholar] [CrossRef] [PubMed]
- Cancer Pain, IASP Website. Available online: https://www.iasp-pain.org/advocacy/global-year/cancer-pain/ (accessed on 20 August 2024).
- Khasabova, I.A.; Stucky, C.L.; Harding-Rose, C.; Eikmeier, L.; Beitz, A.J.; Coicou, L.G.; Hanson, A.E.; Simone, D.A.; Seybold, V.S. Chemical interactions between fibrosarcoma cancer cells and sensory neurons contribute to cancer pain. J. Neurosci. 2007, 27, 10289–10298. [Google Scholar] [CrossRef] [PubMed]
- Falk, S.; Dickenson, A.H. Pain and nociception: Mechanisms of cancer-induced bone pain. J. Clin. Oncol. 2014, 32, 1647–1654. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Oh, J. Neuropathic cancer pain: Prevalence, pathophysiology, and management. Korean J. Intern. Med. 2018, 33, 1058–1069. [Google Scholar] [CrossRef]
- Klose, C.; Straub, I.; Riehle, M.; Ranta, F.; Krautwurst, D.; Ullrich, S.; Meyerhof, W.; Harteneck, C. Fenamates as TRP channel blockers: Mefenamic acid selectively blocks TRPM3. Br. J. Pharmacol. 2011, 162, 1757–1769. [Google Scholar] [CrossRef]
- Cryer, B.; Feldman, M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 413–421. [Google Scholar] [CrossRef]
- Suzuki, H.; Sasaki, E.; Nakagawa, A.; Muraki, Y.; Hatano, N.; Muraki, K. Diclofenac, a nonsteroidal anti-inflammatory drug, is an antagonist of human TRPM3 isoforms. Pharmacol. Res. Perspect. 2016, 4, e00232. [Google Scholar] [CrossRef]
- Biohaven Website. Available online: https://www.biohaven.com/?s=TRPM3 (accessed on 20 August 2024).
- Andrews, M.D.; Af Forselles, K.; Beaumont, K.; Galan, S.R.; Glossop, P.A.; Grenie, M.; Jessiman, A.; Kenyon, A.S.; Lunn, G.; Maw, G.; et al. Discovery of a Selective TRPM8 Antagonist with Clinical Efficacy in Cold-Related Pain. ACS Med. Chem. Lett. 2015, 6, 419–424. [Google Scholar] [CrossRef]
- Horne, D.B.; Biswas, K.; Brown, J.; Bartberger, M.D.; Clarine, J.; Davis, C.D.; Gore, V.K.; Harried, S.; Horner, M.; Kaller, M.R.; et al. Discovery of TRPM8 Antagonist (S)-6-(((3-Fluoro-4-(trifluoromethoxy)phenyl)(3-fluoropyridin-2-yl)methyl)carbamoyl)nicotinic Acid (AMG 333), a Clinical Candidate for the Treatment of Migraine. J. Med. Chem. 2018, 61, 8186–8201. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fiber | Subtype | Positive for |
|---|---|---|
| A-fiber | Proprioceptors |
|
| Aβ-rapid-adapting (RA)—low-threshold mechanoreceptors (LTMRs) |
| |
| Aβ-field/slow-adapting (SA)—LTMRs |
| |
| Aδ-LTMRs |
| |
| Aδ—high-threshold mechanoreceptors (HTMRs) |
| |
| C-fiber | Nociceptors |
|
| Cold thermoreceptors |
| |
| cLTMRs |
| |
| Pruriceptors |
| |
| Atf3 (activating transcription factor 3) cluster expressing injury-induced transcription factors |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandini, M.A.; Zamponi, G.W. Navigating the Controversies: Role of TRPM Channels in Pain States. Int. J. Mol. Sci. 2024, 25, 10284. https://doi.org/10.3390/ijms251910284
Gandini MA, Zamponi GW. Navigating the Controversies: Role of TRPM Channels in Pain States. International Journal of Molecular Sciences. 2024; 25(19):10284. https://doi.org/10.3390/ijms251910284
Chicago/Turabian StyleGandini, Maria A., and Gerald W. Zamponi. 2024. "Navigating the Controversies: Role of TRPM Channels in Pain States" International Journal of Molecular Sciences 25, no. 19: 10284. https://doi.org/10.3390/ijms251910284
APA StyleGandini, M. A., & Zamponi, G. W. (2024). Navigating the Controversies: Role of TRPM Channels in Pain States. International Journal of Molecular Sciences, 25(19), 10284. https://doi.org/10.3390/ijms251910284