
Teosinte-Derived Advanced Backcross Population Harbors Genomic Regions for Grain Yield Attributing Traits in Maize

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
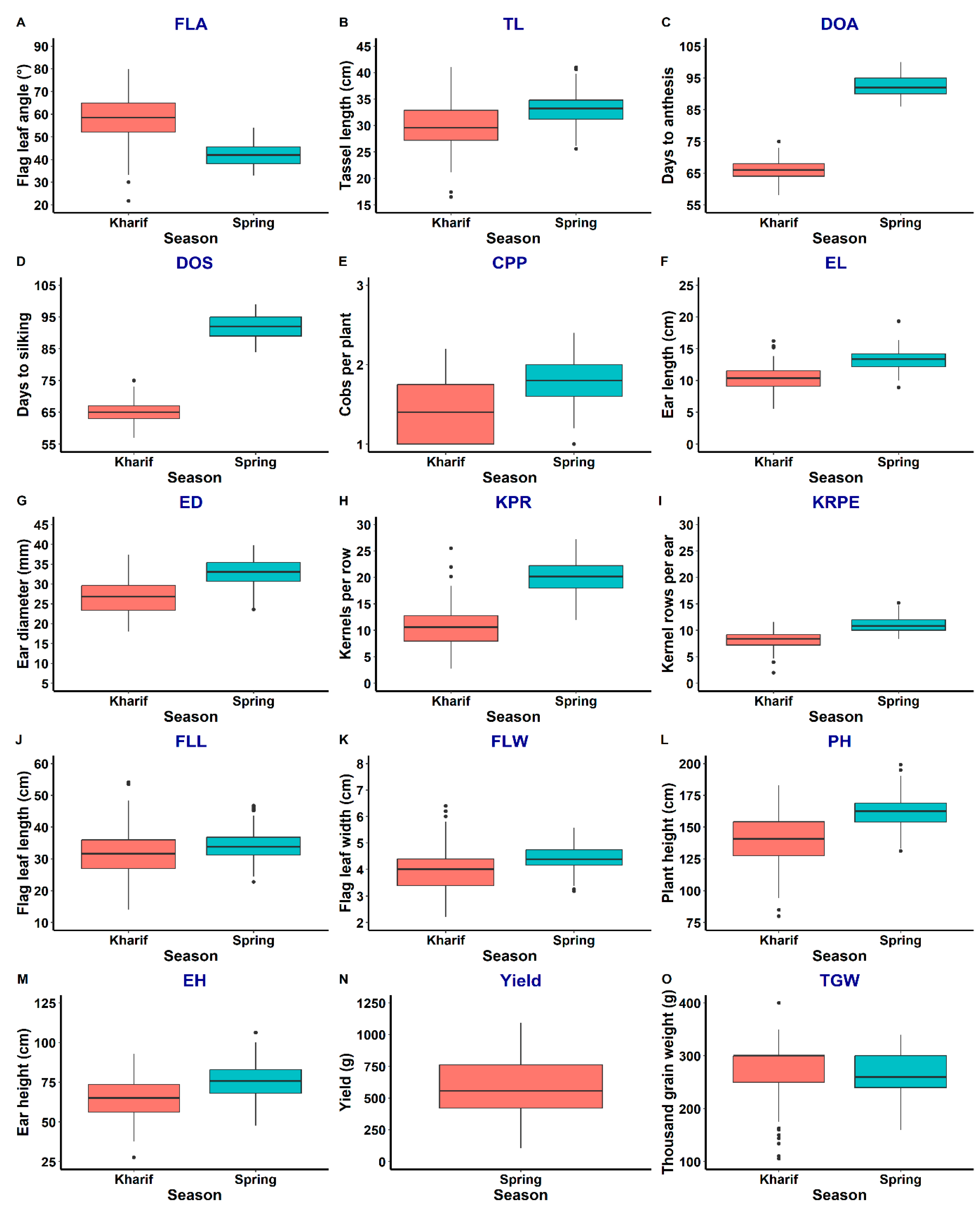
2.1. Evaluation of BC2F2 Population
2.2. QTL Identification
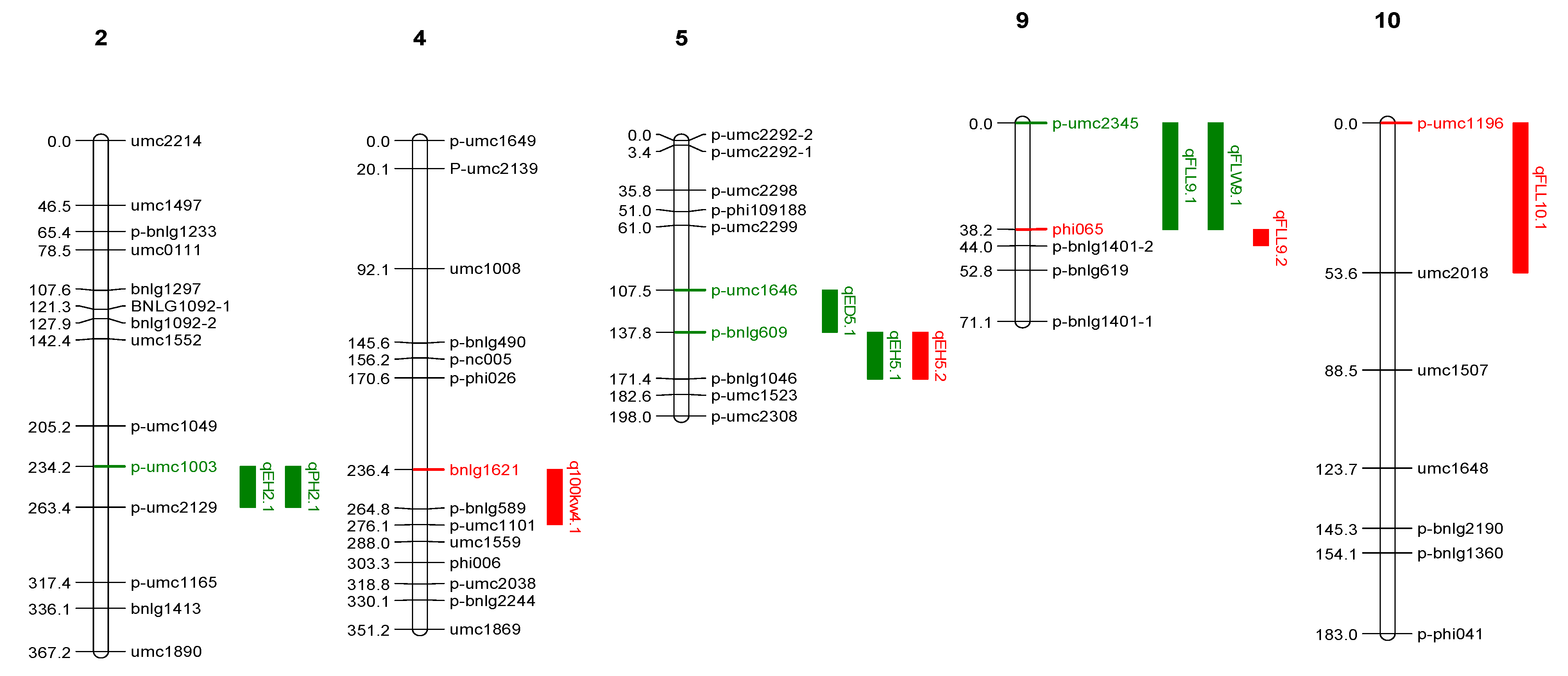
2.2.1. QTLs Identified from LM 13 × Z. parviglumis Derived BC2F2 Families (Population 1)
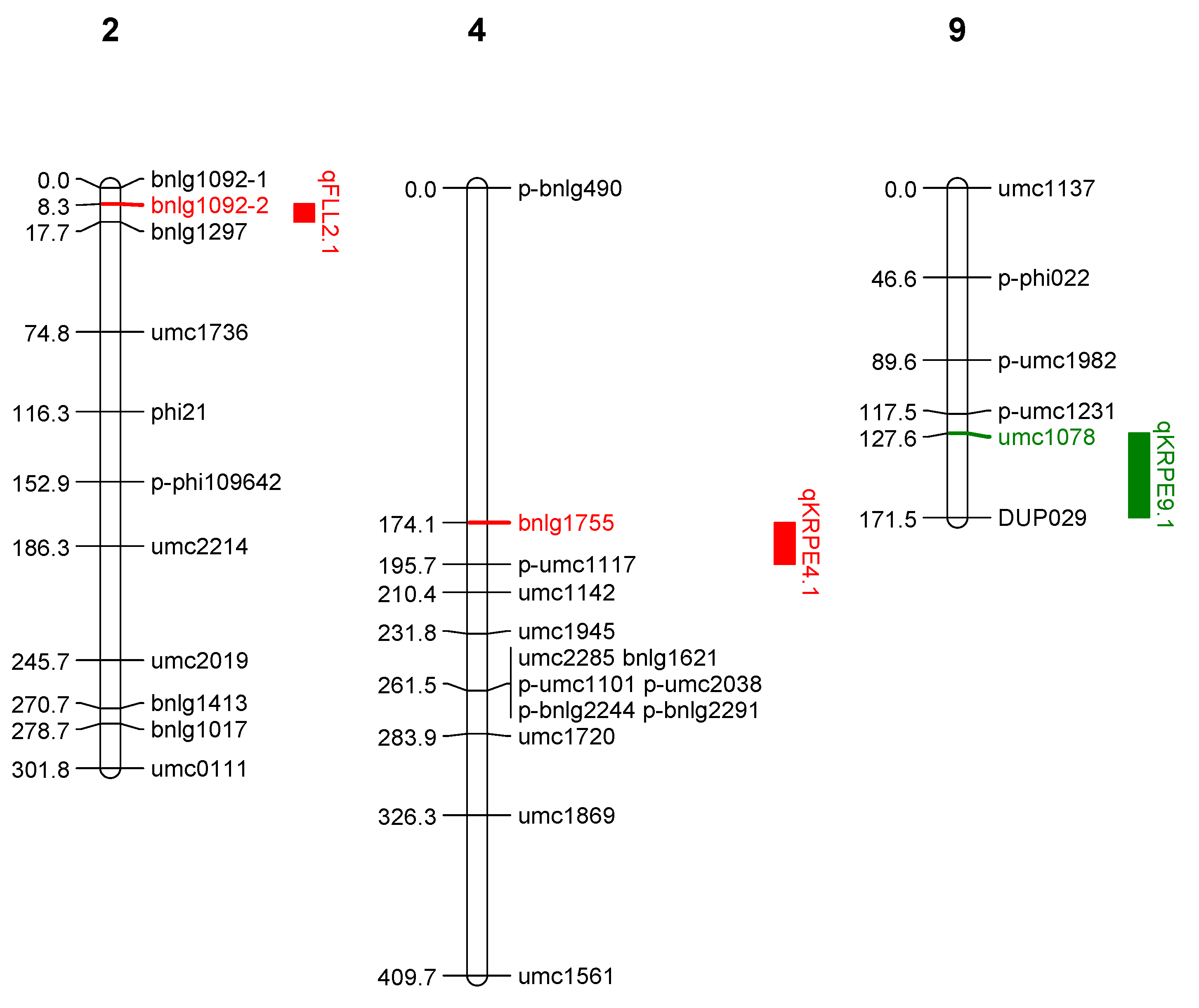
2.2.2. QTLs Identified from LM 14 × Z. parviglumis Derived BC2F2 Families (Population 2)
3. Discussion
3.1. Comparison of QTLs Detected in Both BC2F2 Populations
3.2. Comparison of QTLs Detected in Both BC2F2 Populations and Other Studies in Maize
4. Materials and Methods
4.1. Population Development
4.2. Phenotypic Trait Evaluation
4.3. Correlation and Box Plot Analysis
4.4. Linkage Map Construction and QTLs Mapping
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choudhary, M.; Singh, A.; Gupta, M.; Rakshit, S. Enabling technologies for utilization of maize as a bioenergy feedstock. Biofuels Bioprod. Biorefin. 2019, 14, 402–416. [Google Scholar] [CrossRef]
- Choudhary, M.; Kumar, P.; Kaswan, S.; Jat, S.L. Harnessing the tillering ability of Zea mays ssp. parviglumis in fodder maize breeding. Indian J. Agric. Sci. 2020, 90, 2317–2322. [Google Scholar]
- Joshi, A.; Adhikari, S.; Singh, N.K.; Jaiswal, J.P.; Pant, U.; Singh, R.P.; Pandey, D. Prospecting quantitative trait loci for maydis leaf blight (MLB) resistance using a population of teosinte introgressed maize (Zea mays ssp. mays) and in silico identification of candidate MLB resistance genes. J. Phytopathol. 2023, 171, 118–131. [Google Scholar]
- Keimeso, Z.; Abakemal, D.; Gebreselassie, W. Heterosis and combining ability of highland adapted maize (Zea mays L.) DH lines for desirable agronomic traits. Afr. J. Plant Sci. 2020, 14, 121–133. [Google Scholar]
- Adhikari, S.; Joshi, A.; Kumar, A.; Singh, N.K.; Jaiswal, J.P.; Jeena, A.S.; Pant, U. Developing genetic resources and genetic analysis of plant architecture-related traits in teosinte-introgressed maize popultions. Plant Genet. Resour. 2022, 20, 145–155. [Google Scholar] [CrossRef]
- Ramstein, G.P.; Larsson, S.J.; Cook, J.P.; Edwards, J.W.; Ersoz, E.S.; Flint-Garcia, S.; Gardner, C.A.; Holland, J.B.; Lorenz, A.J.; McMullen, M.D.; et al. Dominance effects and functional enrichments improve prediction of agronomic traits in hybrid maize. Genetics 2020, 215, 215–230. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, N.K.; Adhikari, S.; Joshi, A. Morphological and molecular characterization of teosinte derived maize population. Indian J. Genet. 2019, 79, 670–677. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, N.K.; Jeena, A.S.; Jaiswal, J.P.; Verma, S.S. Evaluation of teosinte derived maize lines for drought tolerance. Plant Genet. Resour. 2020, 33, 60–67. [Google Scholar] [CrossRef]
- Pasztor, K.; Borsos, O. Inheritance and chemical composition in inbred maize (Zea mays L.) 9 teosinte (Zea mays subsp. mexicana (Schrader) Iltis) hybrids. Novenytermeles 1990, 39, 193–213. [Google Scholar]
- Srinivasan, G.; Brewbaker, J.L. Genetic analysis of hybrids between maize and perennial teosinte. II: Ear traits. Maydica 1999, 44, 371–384. [Google Scholar]
- Tanksley, S.D.; Nelson, J.C. Advanced backcross QTL analysis: A method for the simultaneous discovery and transfer of valuable QTLs from unadapted germplasm into elite breeding lines. Theor. Appl. Genet. 1996, 92, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.B.; Song, M.H.; Jin, F.X.; Ahn, S.N.; Suh, J.P.; Hwang, H.G.; McCouch, S.R. Fine mapping of a grain weight quantitative trait locus on rice chromosome 8 using near-isogenic lines derived from a cross between Oryza sativa and Oryza rufipogon. Theor. Appl. Genet. 2007, 113, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chee, P.W. Application of advanced backcross quantitative trait locus (QTL) analysis in crop improvement. J. Plant Breed. Crop Sci. 2010, 2, 221–232. [Google Scholar]
- Sun, Z.; Yin, X.; Ding, J.; Yu, D.; Hu, M.; Sun, X.; Tan, Y.; Sheng, X.; Liu, L.; Mo, Y.; et al. QTL analysis and dissection of panicle components in rice using advanced backcross populations derived from Oryza sativa cultivars HR1128 and ‘Nipponbare’. PLoS ONE 2017, 12, e0175692. [Google Scholar] [CrossRef] [PubMed]
- Sayed, M.A.; Ali, M.B.; Bakry, B.A.; El-Sadek, A.N.; Léon, J. Advanced backcross-quantitative trait loci mapping of grain yield, heading date, and their stability parameters in barley across multienvironmental trials in Egypt. Plant Breed. 2021, 140, 1042–1057. [Google Scholar] [CrossRef]
- Liu, R.; Meng, Q.; Zheng, F.; Kong, L.; Yuan, J.; Lübberstedt, T. Genetic mapping of QTL for maize leaf width combining RIL and IF2 populations. PLoS ONE 2017, 12, e0189441. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Z.; Ding, J.; Wu, Y.; Zhou, B.; Wang, R.; Ma, J.; Wang, S.; Zhang, X.; Xia, Z.; et al. Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize. Front. Plant Sci. 2016, 7, 833. [Google Scholar] [CrossRef]
- Fei, J.; Lu, J.; Jiang, Q.; Liu, Z.; Yao, D.; Qu, J.; Ma, Y. Maize plant architecture trait QTL mapping and candidate gene identification based on multiple environments and double populations. BMC Plant Biol. 2022, 22, 110. [Google Scholar] [CrossRef]
- Choi, J.K.; Sa, K.J.; Park, D.H.; Lim, S.E.; Ryu, S.H.; Park, J.Y.; Park, K.J.; Rhee, H.I.; Lee, M.; Lee, J.K. Construction of genetic linkage map and identification of QTLs related to agronomic traits in DH population of maize (Zea mays L.) using SSR markers. Genes Genom. 2019, 41, 667–678. [Google Scholar] [CrossRef]
- Swamy, B.M.; Kaladhar, K.; Shobha Rani, N.; Prasad, G.S.V.; Viraktamath, B.C.; Reddy, G.A.; Sarla, N. QTL analysis for grain quality traits in 2 BC2F2 populations derived from crosses between Oryza sativa cv Swarna and 2 accessions of O. nivara. J. Hered. 2012, 103, 442–452. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, J. Estimates of marker-associated QTL effects in Monte Carlo backcross generations using multiple regression. Theor. Appl. Genet. 1993, 85, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Beavis, W.D.; Smith, O.S.; Grant, D.M.; Fincher, R.R. Identification of quantitative trait loci using a small sample of top crossed and F4 progeny from maize. Crop Sci. 1994, 34, 882–896. [Google Scholar] [CrossRef]
- Beavis, W.D.; Grant, D.; Albertsen, M.; Fincher, R. Quantitative trait loci for plant height in four maize populations and their associations with qualitative genetic loci. Theor. Appl. Genet. 1991, 83, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Lee, M. DNA markers and plant breeding programs. Adv. Agron. 1995, 55, 265–344. [Google Scholar]
- Koester, R.P.; Sisco, P.H.; Stuber, C.W. Identification of quantitative trait loci controlling days to flowering and plant height in two near isogenic lines of maize. Crop Sci. 1993, 33, 1209–1216. [Google Scholar] [CrossRef]
- Tang, H.; Yan, J.B.; Huang, Y.Q.; Zheng, Y.L.; Li, J.S. QTL mapping of five agronomic traits in maize. Acta. Genet. Sin. 2005, 32, 203–209. [Google Scholar]
- Li, Y.L.; Niu, S.Z.; Dong, Y.B.; Cui, D.Q.; Wang, Y.Z.; Liu, Y.Y.; Wei, M.G. Identification of trait-improving quantitative trait loci for grain yield components from a dent corn inbred line in an advanced backcross BC 2 F 2 population and comparison with its F 2: 3 population in popcorn. Theor. Appl. Genet. 2007, 115, 129–140. [Google Scholar] [CrossRef]
- Adhikari, S.; Joshi, A.; Kumar, A.; Singh, K.N.; Jaiswal, P.J.; Jeena, S.A.; Pant, U. Identification of QTLs for yield and contributing traits in maize-teosinte derived bils under diseased-stressed and control conditions. Genetika 2021, 53, 951–972. [Google Scholar] [CrossRef]
- Su, C.F.; Wang, W.; Gong, S.L.; Zuo, J.H.; Li, S.J.; Xu, S.Z. High density linkage map construction and mapping of yield trait QTLs in maize (Zea mays) using the genotyping-by-sequencing (GBS) technology. Front. Plant Sci. 2017, 8, 706. [Google Scholar] [CrossRef]
- Zhao, Y.M.; Su, C.F. Mapping quantitative trait loci for yield-related traits and predicting candidate genes for grain weight in maize. Sci. Rep. 2019, 9, 16112. [Google Scholar] [CrossRef]
- Jiang, F.Y.; Liu, L.; Li, Z.W.; Bi, Y.Q.; Yin, X.F.; Guo, R.J.; Wang, J.; Zhang, Y.D.; Shaw, R.K.; Fan, X.F. Identification of candidate QTLs and genes for ear diameter by multi-parent population in maize. Genes 2023, 14, 1305. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.P.; Dong, E.F.; Liang, Q.Y.; Bai, Y.; Nan, J.; Yang, Y.; Cai, Y.L. Identification of QTL for fasciated ear related traits in maize. Crop Sci. 2021, 61, 1184–1193. [Google Scholar] [CrossRef]
- Sa, K.J.; Choi, I.Y.; Park, J.Y.; Choi, J.K.; Ryu, S.H.; Lee, J.K. Mapping of QTL for agronomic traits using high-density SNPs with an RIL population in maize. Genes Genom. 2021, 43, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Choudhary, M.; Singh, A.; Sheoran, S.; Kumar, H.; Singla, D.; Rakshit, S. Meta-QTL analysis for mining of candidate genes and constitutive gene network development for viral disease resistance in maize (Zea mays L.). Crop J. 2023, 11, 511–522. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Traits | Chr. | QTLs | LOD | R2 (%) | Linked Markers | Add. Effect |
|---|---|---|---|---|---|---|---|
| Kharif 2020 | |||||||
| 1 | Ear Diameter | 5 | qED5.1 | 3.82 | 12.10 | p-umc1646 & p-bnlg609 | 3.44 |
| 2 | Ear Height | 2 | qEH2.1 | 6.40 | 17.60 | p-umc1003 & p-umc2129 | −12.45 |
| 5 | qEH5.1 | 4.74 | 13.40 | p-bnlg609 & p-bnlg1046 | 19.17 | ||
| 3 | Plant Height | 2 | qPH2.1 | 4.30 | 12.20 | p-umc1049 & p-umc2129 | −17.49 |
| 4 | Flag leaf length | 9 | qFLL9.1 | 4.38 | 12.40 | p-umc2345 & phi065 | −9.56 |
| 5 | Flag leaf width | 9 | qFLW9.1 | 4.05 | 11.50 | p-umc2345 & phi065 | −0.33 |
| Spring 2021 | |||||||
| 6 | 100 kernel weight | 4 | q100kw4.1 | 4.48 | 12.90 | bnlg1621 & p-umc1101 | 1.77 |
| 7 | Ear height | 5 | qEH5.2 | 4.82 | 13.90 | p-umc609 & p-bnlg1046 | 8.62 |
| 8 | Flag leaf length | 9 | qFLL9.2 | 4.13 | 12.00 | phi065 & p-bnlg1401-2 | 10.92 |
| 10 | qFLL10.1 | 3.68 | 10.80 | umc1196 & umc2018 | −3.00 | ||
| S. No. | Traits | Chr. | QTLs | LOD | R2 (%) | Linked Markers | Add. Effect |
|---|---|---|---|---|---|---|---|
| Kharif 2020 | |||||||
| 1 | Kernel rows per ear | 9 | qKRPE9.1 | 3.70 | 13.70 | umc1078 & DUP029 | −8.33 |
| Spring 2021 | |||||||
| 2 | Flag leaf length | 2 | qFLL2.1 | 3.64 | 10.30 | bnlg1092-2 & bnlg1297 | 2.61 |
| 3 | Kernel rows per ear | 4 | qKRPE4.1 | 5.09 | 14.60 | bnlg1755 & pumc1117 | 7.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Choudhary, M.; Sheoran, S.; Longmei, N.; Kumar, B.; Jat, B.S.; Dagla, M.C.; Bhushan, B.; Aggarwal, S.K.; Bagaria, P.K.; et al. Teosinte-Derived Advanced Backcross Population Harbors Genomic Regions for Grain Yield Attributing Traits in Maize. Int. J. Mol. Sci. 2024, 25, 10300. https://doi.org/10.3390/ijms251910300
Kumar P, Choudhary M, Sheoran S, Longmei N, Kumar B, Jat BS, Dagla MC, Bhushan B, Aggarwal SK, Bagaria PK, et al. Teosinte-Derived Advanced Backcross Population Harbors Genomic Regions for Grain Yield Attributing Traits in Maize. International Journal of Molecular Sciences. 2024; 25(19):10300. https://doi.org/10.3390/ijms251910300
Chicago/Turabian StyleKumar, Pardeep, Mukesh Choudhary, Seema Sheoran, Ningthai Longmei, Bhupender Kumar, Bahadur Singh Jat, Manesh Chander Dagla, Bharat Bhushan, Sumit Kumar Aggarwal, Pravin Kumar Bagaria, and et al. 2024. "Teosinte-Derived Advanced Backcross Population Harbors Genomic Regions for Grain Yield Attributing Traits in Maize" International Journal of Molecular Sciences 25, no. 19: 10300. https://doi.org/10.3390/ijms251910300