
Advances in Chiral Pincer Complexes: Insights and Applications in Catalytic Asymmetric Reactions †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Enantioselective Hydrogenation of C=O Bonds
2.1. Asymmetric Hydrogenation
2.2. Asymmetric Transfer Hydrogenation
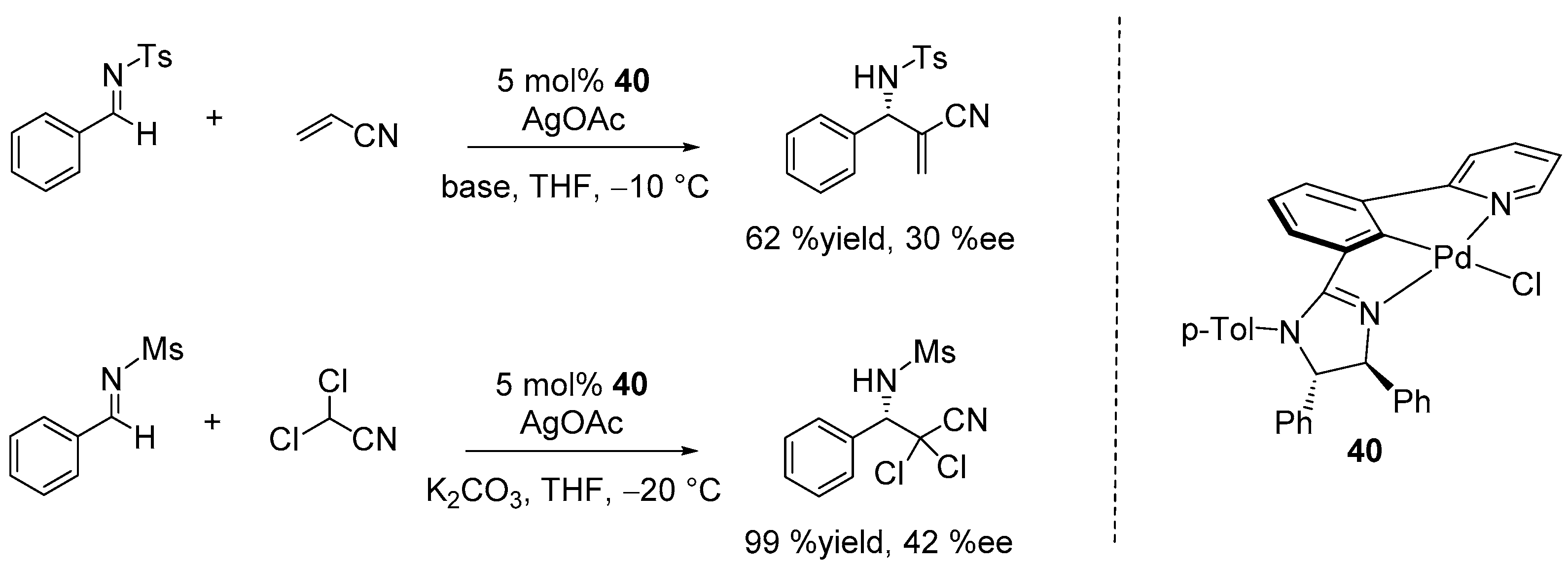
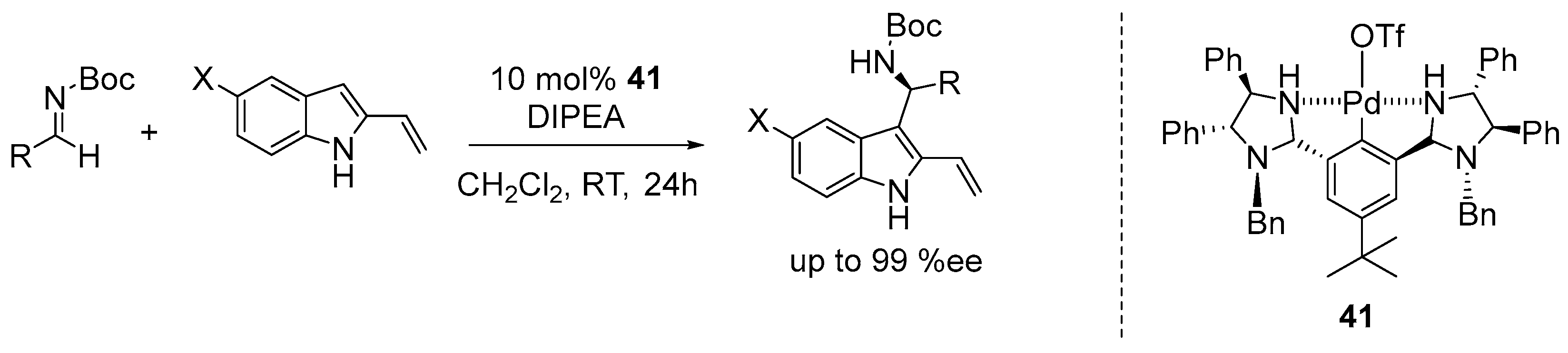
3. Enantioselective Hydrogenation of C=N Bonds
4. Enantioselective Hydrogenation of C=C Bonds
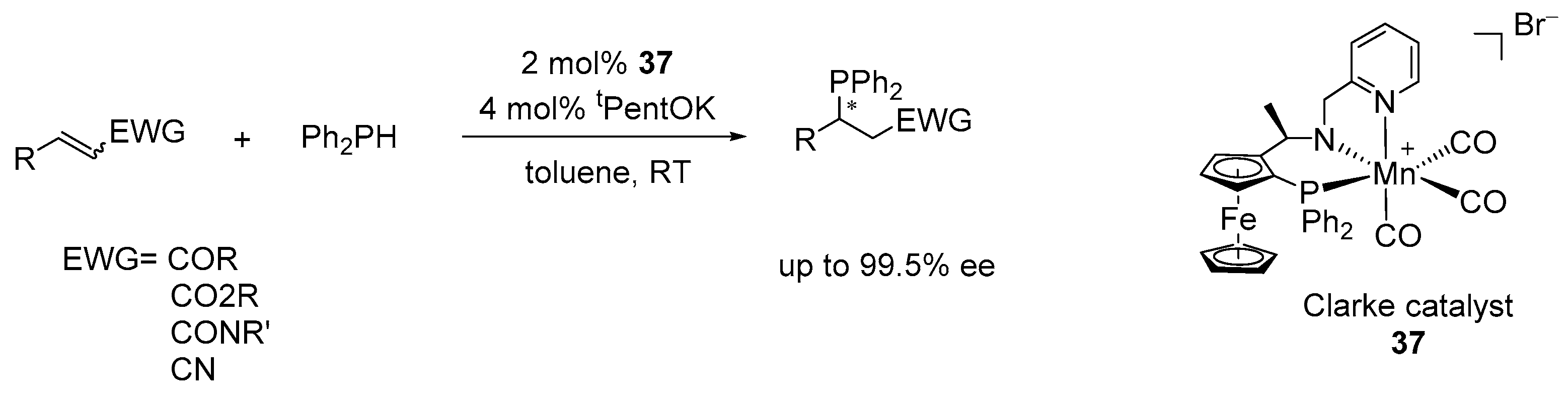
5. Enantioselective Hydrophosphination
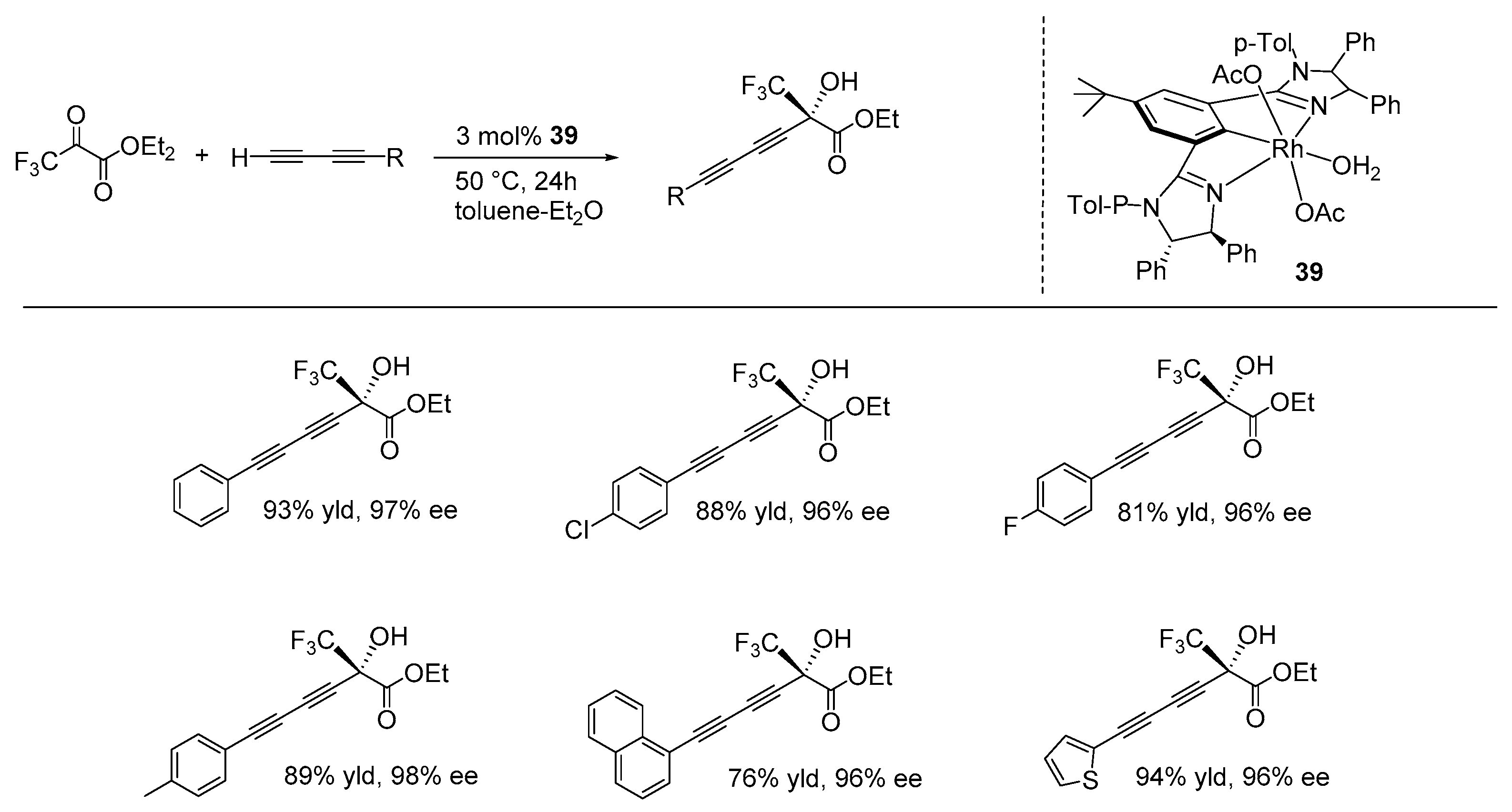
6. Enantioselective Alkynylation
7. Miscellaneous Enantioselective Reactions
8. Conclusions
Funding
Conflicts of Interest
References
- Lawrence, M.A.W.; Green, K.-A.; Nelson, P.N.; Lorraine, S.C. Review: Pincer ligands—Tunable, versatile and applicable. Polyhedron 2018, 143, 11–27. [Google Scholar] [CrossRef]
- Gelman, D.; Musa, S. Coordination Versatility of sp3-Hybridized Pincer Ligands toward Ligand–Metal Cooperative Catalysis. ACS Catal. 2012, 2, 2456–2466. [Google Scholar] [CrossRef]
- Albrecht, M.; van Koten, G. Platinum Group Organometallics Based on “Pincer” Complexes: Sensors, Switches, and Catalysts. Angew. Chem. Int. Ed. 2001, 40, 3750–3781. [Google Scholar] [CrossRef]
- Hao, X.; Niu, J.; Zhao, X.; Gong, J.; Song, M. Development of Pincer Catalysts with Selected Group 8~10 Metals. Chin. J. Org. Chem. 2013, 33, 663–674. [Google Scholar] [CrossRef]
- Chase, P.A.; Gossage, R.A.; van Koten, G. Modern Organometallic Multidentate Ligand Design Strategies: The Birth of the Privileged “Pincer” Ligand Platform. In The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications; van Koten, G., Gossage, R.A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–15. [Google Scholar]
- Richards, C.J.; Fossey, J.S. CHAPTER 3—Chiral pincer complexes and their application to asymmetric synthesis. In The Chemistry of Pincer Compounds; Morales-Morales, D., Jensen, C.M., Eds.; Elsevier Science B.V.: Amsterdam, The Netherland, 2007; pp. 45–77. [Google Scholar]
- Ito, J.-I.; Nishiyama, H. Chapter 1—Chiral Pincer Complexes for Asymmetric Reactions. In Pincer Compounds; Morales-Morales, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–18. [Google Scholar]
- Wang, H.; Wen, J.; Zhang, X. Chiral Tridentate Ligands in Transition Metal-Catalyzed Asymmetric Hydrogenation. Chem. Rev. 2021, 121, 7530–7567. [Google Scholar] [CrossRef]
- Chakrabortty, S.; de Bruin, B.; de Vries, J.G. Cobalt-Catalyzed Asymmetric Hydrogenation: Substrate Specificity and Mechanistic Variability. Angew. Chem. Int. Ed. 2024, 63, e202315773. [Google Scholar] [CrossRef]
- Ito, J.-I.; Asai, R.; Nishiyama, H. Asymmetric Direct Alkynylation Catalyzed by Chiral Ru−Bis(oxazolinyl)phenyl Complexes. Org. Lett. 2010, 12, 3860–3862. [Google Scholar] [CrossRef]
- Shiomi, T.; Adachi, T.; Toribatake, K.; Zhou, L.; Nishiyama, H. Asymmetric β-boration of α,β-unsaturated carbonyl compounds promoted by chiral rhodium–bisoxazolinylphenyl catalysts. Chem. Commun. 2009, 40, 5987–5989. [Google Scholar] [CrossRef]
- Coeffard, V.; Aylward, M.; Guiry, P.J. First Regio- and Enantioselective Chromium-Catalyzed Homoallenylation of Aldehydes. Angew. Chem. Int. Ed. 2009, 48, 9152–9155. [Google Scholar] [CrossRef]
- Inoue, M.; Suzuki, T.; Nakada, M. Asymmetric catalysis of Nozaki-Hiyama allylation and methallylation with a new tridentate bis(oxazolinyl)carbazole ligand. J. Am. Chem. Soc. 2003, 125, 1140–1141. [Google Scholar] [CrossRef]
- Inoue, H.; Kikuchi, M.; Ito, J.-I.; Nishiyama, H. Chiral Phebox–rhodium complexes as catalysts for asymmetric direct aldol reaction. Tetrahedron 2008, 64, 493–499. [Google Scholar] [CrossRef]
- Mizuno, M.; Inoue, H.; Naito, T.; Zhou, L.; Nishiyama, H. Asymmetric, Regioselective Direct Aldol Coupling of Enones and Aldehydes with Chiral Rhodium(bis-oxazolinylphenyl) Catalysts. Chem. Eur. J. 2009, 15, 8985–8988. [Google Scholar] [CrossRef] [PubMed]
- Postolache, R.; Pérez, J.M.; Castiñeira Reis, M.; Ge, L.; Sinnema, E.G.; Harutyunyan, S.R. Manganese(I)-Catalyzed Asymmetric Hydrophosphination of α,β-Unsaturated Carbonyl Derivatives. Org. Lett. 2023, 25, 1611–1615. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.-H.; Melen, R.L.; Gade, L.H. Anionic Chiral Tridentate N-Donor Pincer Ligands in Asymmetric Catalysis. Acc. Chem. Res. 2014, 47, 3162–3173. [Google Scholar] [CrossRef]
- Nakamura, S.; Hyodo, K.; Nakamura, M.; Nakane, D.; Masuda, H. Catalytic Enantioselective Allylation of Ketimines by Using Palladium Pincer Complexes with Chiral Bis(imidazoline)s. Chem. Eur. J. 2013, 19, 7304–7309. [Google Scholar] [CrossRef]
- Gademann, K.; Chavez, D.E.; Jacobsen, E.N. Highly Enantioselective Inverse-Electron-Demand Hetero-Diels–Alder Reactions of α,β-Unsaturated Aldehydes. Angew. Chem. Int. Ed. 2002, 41, 3059–3061. [Google Scholar] [CrossRef]
- Arai, T.; Kakino, J. Catalytic Asymmetric Synthesis of 3-Indolyl Methanamines Using Unprotected Indoles and N-Boc Imines under Basic Conditions. Angew. Chem. Int. Ed. 2016, 55, 15263–15267. [Google Scholar] [CrossRef]
- Wen, J.; Wang, F.; Zhang, X. Asymmetric hydrogenation catalyzed by first-row transition metal complexes. Chem. Soc. Rev. 2021, 50, 3211–3237. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, T.; Ooka, H.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Practical Enantioselective Hydrogenation of Aromatic Ketones. J. Am. Chem. Soc. 1995, 117, 2675–2676. [Google Scholar] [CrossRef]
- Doucet, H.; Ohkuma, T.; Murata, K.; Yokozawa, T.; Kozawa, M.; Katayama, E.; England, A.F.; Ikariya, T.; Noyori, R. trans-[RuCl2(phosphane)2(1,2-diamine)] and Chiral trans-[RuCl2(diphosphane)(1,2-diamine)]: Shelf-Stable Precatalysts for the Rapid, Productive, and Stereoselective Hydrogenation of Ketones. Angew. Chem. Int. Ed. 1998, 37, 1703–1707. [Google Scholar] [CrossRef]
- Ohkuma, T.; Koizumi, M.; Doucet, H.; Pham, T.; Kozawa, M.; Murata, K.; Katayama, E.; Yokozawa, T.; Ikariya, T.; Noyori, R. Asymmetric Hydrogenation of Alkenyl, Cyclopropyl, and Aryl Ketones. RuCl2(xylbinap)(1,2-diamine) as a Precatalyst Exhibiting a Wide Scope. J. Am. Chem. Soc. 1998, 120, 13529–13530. [Google Scholar] [CrossRef]
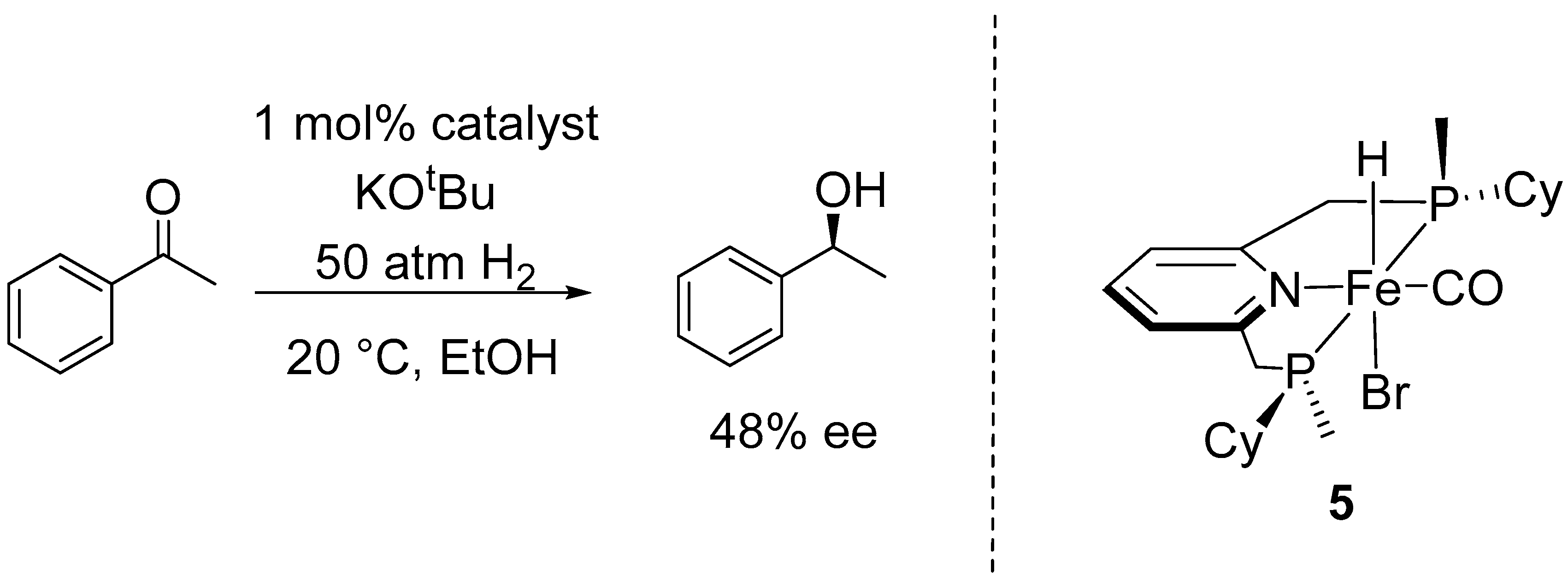
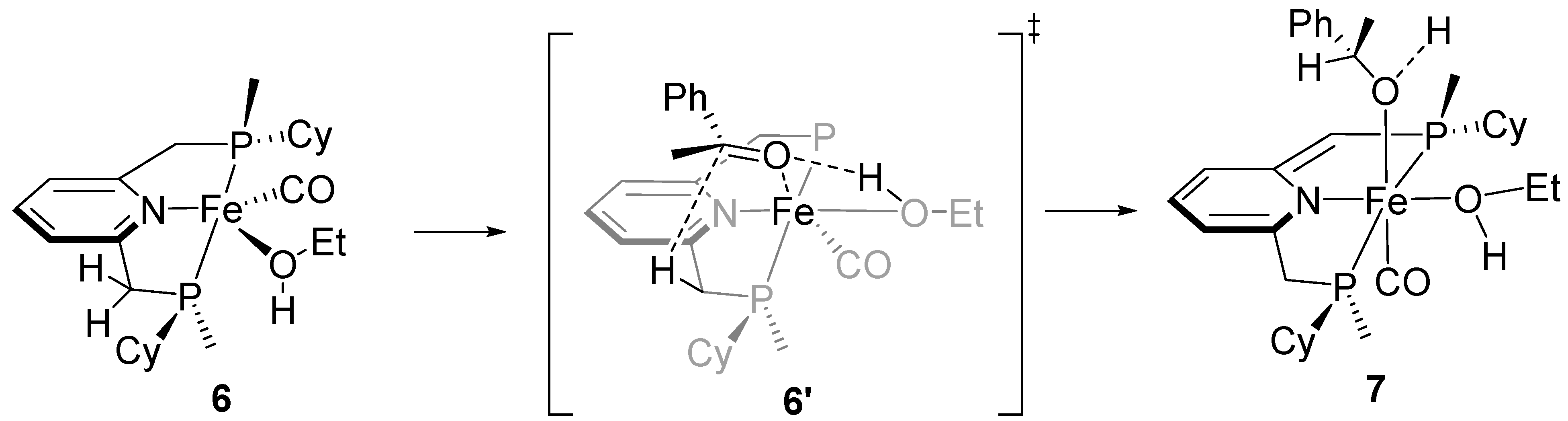
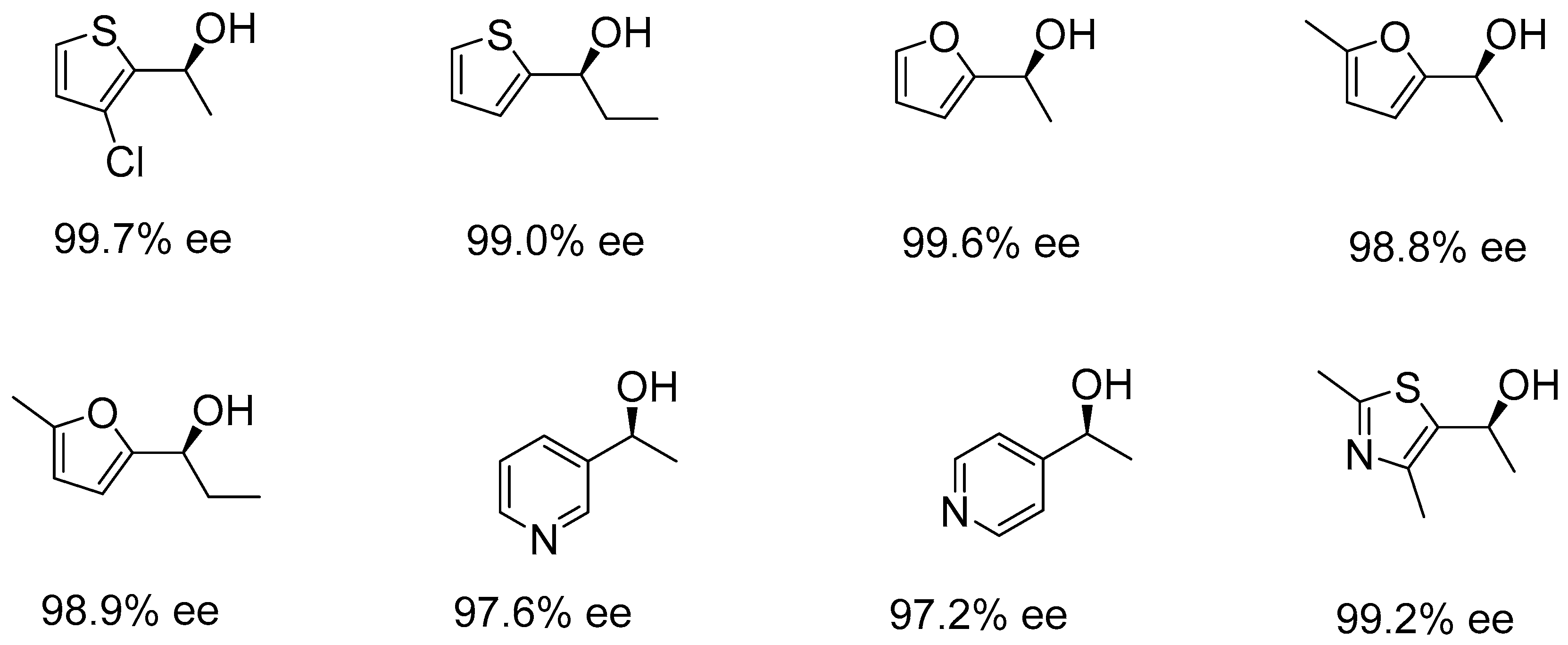
- Huber, R.; Passera, A.; Mezzetti, A. Iron(II)-Catalyzed Hydrogenation of Acetophenone with a Chiral, Pyridine-Based PNP Pincer Ligand: Support for an Outer-Sphere Mechanism. Organometallics 2018, 37, 396–405. [Google Scholar] [CrossRef]
- Widegren, M.B.; Harkness, G.J.; Slawin, A.M.Z.; Cordes, D.B.; Clarke, M.L. A Highly Active Manganese Catalyst for Enantioselective Ketone and Ester Hydrogenation. Angew. Chem. Int. Ed. 2017, 56, 5825–5828. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Passera, A.; Gubler, E.; Mezzetti, A. P-Stereogenic PN(H)P Iron(II) Catalysts for the Asymmetric Hydrogenation of Ketones: The Importance of Non-Covalent Interactions in Rational Ligand Design by Computation. Adv. Synth. Catal. 2018, 360, 2900–2913. [Google Scholar] [CrossRef]
- Császár, Z.; Pőrgye, Z.E.; Tóth-Farsang, E.; Kovács, M.; Bényei, A.C.; Bakos, J.; Farkas, G. Ruthenium complexes of new chiral phosphine-amine-ether ligands (Ru-PNO) for asymmetric hydrogenation—the role of backbone chirality in pincer ligand design. Appl. Organomet. Chem. 2024, 38, e7379. [Google Scholar] [CrossRef]
- Lagaditis, P.O.; Sues, P.E.; Sonnenberg, J.F.; Wan, K.Y.; Lough, A.J.; Morris, R.H. Iron(II) Complexes Containing Unsymmetrical P–N–P′ Pincer Ligands for the Catalytic Asymmetric Hydrogenation of Ketones and Imines. J. Am. Chem. Soc. 2014, 136, 1367–1380. [Google Scholar] [CrossRef]
- Sonnenberg, J.F.; Lough, A.J.; Morris, R.H. Synthesis of Iron P-N-P′ and P-NH-P′ Asymmetric Hydrogenation Catalysts. Organometallics 2014, 33, 6452–6465. [Google Scholar] [CrossRef]
- Sonnenberg, J.F.; Wan, K.Y.; Sues, P.E.; Morris, R.H. Ketone Asymmetric Hydrogenation Catalyzed by P-NH-P′ Pincer Iron Catalysts: An Experimental and Computational Study. ACS Catal. 2017, 7, 316–326. [Google Scholar] [CrossRef]
- Zirakzadeh, A.; Kirchner, K.; Roller, A.; Stöger, B.; Widhalm, M.; Morris, R.H. Iron(II) Complexes Containing Chiral Unsymmetrical PNP′ Pincer Ligands: Synthesis and Application in Asymmetric Hydrogenations. Organometallics 2016, 35, 3781–3787. [Google Scholar] [CrossRef]
- Smith, S.A.M.; Lagaditis, P.O.; Lüpke, A.; Lough, A.J.; Morris, R.H. Unsymmetrical Iron P-NH-P′ Catalysts for the Asymmetric Pressure Hydrogenation of Aryl Ketones. Chem. Eur. J. 2017, 23, 7212–7216. [Google Scholar] [CrossRef]
- Langer, R.; Iron, M.A.; Konstantinovski, L.; Diskin-Posner, Y.; Leitus, G.; Ben-David, Y.; Milstein, D. Iron Borohydride Pincer Complexes for the Efficient Hydrogenation of Ketones under Mild, Base-Free Conditions: Synthesis and Mechanistic Insight. Chem. Eur. J. 2012, 18, 7196–7209. [Google Scholar] [CrossRef] [PubMed]
- Langer, R.; Leitus, G.; Ben-David, Y.; Milstein, D. Efficient Hydrogenation of Ketones Catalyzed by an Iron Pincer Complex. Angew. Chem. Int. Ed. 2011, 50, 2120–2124. [Google Scholar] [CrossRef]
- Samec, J.S.M.; Bäckvall, J.-E.; Andersson, P.G.; Brandt, P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006, 35, 237–248. [Google Scholar] [CrossRef]
- Haack, K.-J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. The Catalyst Precursor, Catalyst, and Intermediate in the RuII-Promoted Asymmetric Hydrogen Transfer between Alcohols and Ketones. Angew. Chem. Int. Ed. 1997, 36, 285–288. (In English) [Google Scholar] [CrossRef]
- Dub, P.A.; Gordon, J.C. Metal–Ligand Bifunctional Catalysis: The “Accepted” Mechanism, the Issue of Concertedness, and the Function of the Ligand in Catalytic Cycles Involving Hydrogen Atoms. ACS Catal. 2017, 7, 6635–6655. [Google Scholar] [CrossRef]
- Garbe, M.; Junge, K.; Walker, S.; Wei, Z.; Jiao, H.; Spannenberg, A.; Bachmann, S.; Scalone, M.; Beller, M. Manganese(I)-Catalyzed Enantioselective Hydrogenation of Ketones Using a Defined Chiral PNP Pincer Ligand. Angew. Chem. Int. Ed. 2017, 56, 11237–11241. [Google Scholar] [CrossRef]
- Garbe, M.; Wei, Z.; Tannert, B.; Spannenberg, A.; Jiao, H.; Bachmann, S.; Scalone, M.; Junge, K.; Beller, M. Enantioselective Hydrogenation of Ketones using Different Metal Complexes with a Chiral PNP Pincer Ligand. Adv. Synth. Catal. 2019, 361, 1913–1920. [Google Scholar] [CrossRef]
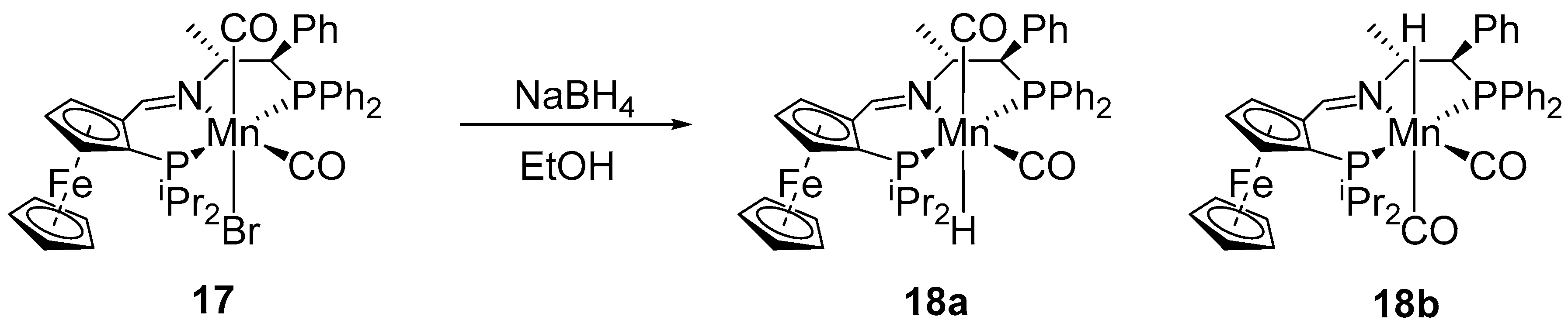
- Seo, C.S.G.; Tsui, B.T.H.; Gradiski, M.V.; Smith, S.A.M.; Morris, R.H. Enantioselective direct, base-free hydrogenation of ketones by a manganese amido complex of a homochiral, unsymmetrical P–N–P′ ligand. Catal. Sci. Technol. 2021, 11, 3153–3163. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Q.; Li, L.; Jiang, J.; Sun, H.; Li, L.; Liu, T.; Zhang, L.; Li, C. Ruthenium-catalyzed asymmetric hydrogenation of aromatic and heteroaromatic ketones using cinchona alkaloid-derived NNP ligands. RSC Adv. 2022, 12, 14912–14916. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Chen, Q.; Li, L.; Jiang, J.; Sun, H.; Zhao, C.; Yang, Y.; Li, C. Cinchona-Alkaloid-Derived NNP Ligand for Iridium-Catalyzed Asymmetric Hydrogenation of Ketones. Org. Lett. 2022, 24, 415–419. [Google Scholar] [CrossRef]
- Palmer, M.J.; Wills, M. Asymmetric transfer hydrogenation of C=O and C=N bonds. Tetrahedron Asymmetry 1999, 10, 2045–2061. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Lan, S.; Chen, T.; Luo, H.; Zhu, L.; Chen, N.; Liu, J.; Yang, S.; Cotman, A.E.; Zhang, Q.; et al. Catalytic Asymmetric Transfer Hydrogenation of Acylboronates: BMIDA as the Privileged Directing Group. J. Am. Chem. Soc. 2024, 146, 20357–20369. [Google Scholar] [CrossRef]
- Dub, P.A.; Tkachenko, N.V.; Vyas, V.K.; Wills, M.; Smith, J.S.; Tretiak, S. Enantioselectivity in the Noyori–Ikariya Asymmetric Transfer Hydrogenation of Ketones. Organometallics 2021, 40, 1402–1410. [Google Scholar] [CrossRef]
- Zirakzadeh, A.; de Aguiar, S.R.M.M.; Stöger, B.; Widhalm, M.; Kirchner, K. Enantioselective Transfer Hydrogenation of Ketones Catalyzed by a Manganese Complex Containing an Unsymmetrical Chiral PNP′ Tridentate Ligand. ChemCatChem 2017, 9, 1744–1748. [Google Scholar] [CrossRef]
- Demmans, K.Z.; Olson, M.E.; Morris, R.H. Asymmetric Transfer Hydrogenation of Ketones with Well-Defined Manganese(I) PNN and PNNP Complexes. Organometallics 2018, 37, 4608–4618. [Google Scholar] [CrossRef]
- Yang, J.; Yao, L.; Wang, Z.; Zuo, Z.; Liu, S.; Gao, P.; Han, M.; Liu, Q.; Solan, G.A.; Sun, W.-H. Manganese(I)-catalyzed asymmetric (transfer) hydrogenation of ketones: An insight into the effect of chiral PNN and NN ligands. J. Catal. 2023, 418, 40–50. [Google Scholar] [CrossRef]
- Zhang, S.; Ma, Z.; Li, Y.; Su, Y.; Ma, N.; Guo, X.; Li, L.; Liu, Q.; Wang, Z. Enhancing enantioselectivity of manganese catalyst for asymmetric transfer hydrogenation of ketones through P,N,N-chelation of a cyclooctyl pyridine. J. Catal. 2024, 437, 115682. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, S.; Ma, Z.; Li, L.; Yan, X.; Cao, Q.; Su, Y.; Ma, N.; Wang, Z. Cycloheptyl-fused-PNN-manganese catalyzed asymmetric transfer hydrogenation of ketones. Mol. Catal. 2024, 564, 114274. [Google Scholar] [CrossRef]
- Xu, X.; You, Y.; Jin, M.Y.; Meng, F.-J.; Xu, C.; Xing, X. Pincer Ru with a single stereogenic identity for highly efficient asymmetric transfer hydrogenation of ketones. Sci. China Chem. 2023, 66, 1443–1449. [Google Scholar] [CrossRef]
- Chelucci, G.; Baldino, S.; Baratta, W. Ruthenium and osmium complexes containing 2-(aminomethyl)pyridine (Ampy)-based ligands in catalysis. Coord. Chem. Rev. 2015, 300, 29–85. [Google Scholar] [CrossRef]
- Baratta, W.; Chelucci, G.; Magnolia, S.; Siega, K.; Rigo, P. Highly Productive CNN Pincer Ruthenium Catalysts for the Asymmetric Reduction of Alkyl Aryl Ketones. Chem. Eur. J. 2009, 15, 726–732. [Google Scholar] [CrossRef]
- Cabré, A.; Verdaguer, X.; Riera, A. Recent Advances in the Enantioselective Synthesis of Chiral Amines via Transition Metal-Catalyzed Asymmetric Hydrogenation. Chem. Rev. 2022, 122, 269–339. [Google Scholar] [CrossRef] [PubMed]
- Foubelo, F.; Nájera, C.; Retamosa, M.G.; Sansano, J.M.; Yus, M. Catalytic asymmetric synthesis of 1,2-diamines. Chem. Soc. Rev. 2024, 53, 7983–8085. [Google Scholar] [CrossRef]
- Liu, Y.; Yue, X.; Li, L.; Li, Z.; Zhang, L.; Pu, M.; Yang, Z.; Wang, C.; Xiao, J.; Lei, M. Asymmetric Induction with a Chiral Amine Catalyzed by a Ru-PNP Pincer Complex: Insight from Theoretical Investigation. Inorg. Chem. 2020, 59, 8404–8411. [Google Scholar] [CrossRef]
- Sablong, R.; Osborn, J.A. The asymmetric hydrogenation of imines using tridentate C2 diphosphine complexes of iridium(I) and rhodium(I). Tetrahedron Lett. 1996, 37, 4937–4940. [Google Scholar] [CrossRef]
- Kutlescha, K.; Irrgang, T.; Kempe, R. Novel Amido-Complexes for the Efficient Asymmetric Hydrogenation of Imines. Adv. Synth. Catal. 2010, 352, 3126–3130. [Google Scholar] [CrossRef]
- Menéndez-Pedregal, E.; Vaquero, M.; Lastra, E.; Gamasa, P.; Pizzano, A. Highly Enantioselective Hydrogenation of N-Aryl Imines Derived from Acetophenones by Using Ru–Pybox Complexes under Hydrogenation or Transfer Hydrogenation Conditions in Isopropanol. Chem. Eur. J. 2015, 21, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Seo, C.S.G.; Tannoux, T.; Smith, S.A.M.; Lough, A.J.; Morris, R.H. Enantioselective Hydrogenation of Activated Aryl Imines Catalyzed by an Iron(II) P-NH-P′ Complex. J. Org. Chem. 2019, 84, 12040–12049. [Google Scholar] [CrossRef]
- Wang, M.; Liu, S.; Liu, H.; Wang, Y.; Lan, Y.; Liu, Q. Asymmetric hydrogenation of ketimines with minimally different alkyl groups. Nature 2024, 631, 556–562. [Google Scholar] [CrossRef]
- Osborn, J.A.; Jardine, F.H.; Young, J.F.; Wilkinson, G. The preparation and properties of tris(triphenylphosphine)halogenorhodium(I) and some reactions thereof including catalytic homogeneous hydrogenation of olefins and acetylenes and their derivatives. J. Chem. Soc. A Inorg. Phys. Theor. 1966, 1711–1732. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric Hydrogenations (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 1998–2007. [Google Scholar] [CrossRef]
- Knowles, W.S.; Sabacky, M.J. Catalytic asymmetric hydrogenation employing a soluble, optically active, rhodium complex. Chem. Commun. 1968, 1445–1446. [Google Scholar] [CrossRef]
- Horner, L.; Büthe, H.; Siegel, H. Hydrierung und isomerisierung von olefinen mit homogen gelösten phosphin-rhodium-komplexen. Tetrahedron Lett. 1968, 9, 4023–4026. [Google Scholar] [CrossRef]
- Bianchini, C.; Mantovani, G.; Meli, A.; Migliacci, F.; Zanobini, F.; Laschi, F.; Sommazzi, A. Oligomerisation of Ethylene to Linear α-Olefins by new C- and C-Symmetric [2,6-Bis(imino)pyridyl]iron and -cobalt Dichloride Complexes. Eur. J. Inorg. Chem. 2003, 2003, 1620–1631. [Google Scholar] [CrossRef]
- Monfette, S.; Turner, Z.R.; Semproni, S.P.; Chirik, P.J. Enantiopure C1-Symmetric Bis(imino)pyridine Cobalt Complexes for Asymmetric Alkene Hydrogenation. J. Am. Chem. Soc. 2012, 134, 4561–4564. [Google Scholar] [CrossRef]
- Friedfeld, M.R.; Shevlin, M.; Margulieux, G.W.; Campeau, L.-C.; Chirik, P.J. Cobalt-Catalyzed Enantioselective Hydrogenation of Minimally Functionalized Alkenes: Isotopic Labeling Provides Insight into the Origin of Stereoselectivity and Alkene Insertion Preferences. J. Am. Chem. Soc. 2016, 138, 3314–3324. [Google Scholar] [CrossRef]
- Viereck, P.; Krautwald, S.; Pabst, T.P.; Chirik, P.J. A Boron Activating Effect Enables Cobalt-Catalyzed Asymmetric Hydrogenation of Sterically Hindered Alkenes. J. Am. Chem. Soc. 2020, 142, 3923–3930. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Z.; Leng, X.; Zhu, H.; Liu, G.; Huang, Z. Transfer Hydrogenation of Alkenes Using Ethanol Catalyzed by a NCP Pincer Iridium Complex: Scope and Mechanism. J. Am. Chem. Soc. 2018, 140, 4417–4429. [Google Scholar] [CrossRef]
- Liu, F.; Goldman, S.A. Efficient thermochemical alkane dehydrogenation and isomerization catalyzed by an iridium pincer complex. Chem. Commun. 1999, 655–656. [Google Scholar] [CrossRef]
- Gupta, M.; Hagen, C.; Kaska, W.C.; Cramer, R.E.; Jensen, C.M. Catalytic Dehydrogenation of Cycloalkanes to Arenes by a Dihydrido Iridium P−C−P Pincer Complex. J. Am. Chem. Soc. 1997, 119, 840–841. [Google Scholar] [CrossRef]
- Qian, L.; Tang, X.; Huang, Z.; Wang, Y.; Liu, G.; Huang, Z. Chiral Iridium Complexes of Anionic NCP Pincer Ligand for Asymmetric Transfer Hydrogenation of 1,1-Diarylethenes with Ethanol. Org. Lett. 2021, 23, 8978–8983. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Qian, L.; Liu, G.; Huang, Z. Iridium-Catalyzed Asymmetric Transfer Hydrogenation of 1-Aryl-1-alkylethenes with Ethanol. Org. Lett. 2023, 25, 4950–4954. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Yu, C.; Gan, L.; Tang, X.; Wang, Y.; Liu, G.; Leng, X.; Sun, Z.; Guo, Y.; Xue, X.-S.; et al. Iridium-Catalyzed Enantioselective Transfer Hydrogenation of 1,1-Dialkylethenes with Ethanol: Scope and Mechanism. J. Am. Chem. Soc. 2024, 146, 3427–3437. [Google Scholar] [CrossRef]
- Zhang, X.-X.; Zhang, Y.; Liao, L.; Gao, Y.; Su, H.E.M.; Yu, J.-S. Catalytic Asymmetric Isomerization of (Homo)Allylic Alcohols: Recent Advances and Challenges. ChemCatChem 2022, 14, e202200126. [Google Scholar] [CrossRef]
- Clevenger, A.L.; Stolley, R.M.; Aderibigbe, J.; Louie, J. Trends in the Usage of Bidentate Phosphines as Ligands in Nickel Catalysis. Chem. Rev. 2020, 120, 6124–6196. [Google Scholar] [CrossRef] [PubMed]
- Belkova, N.V.; Filippov, O.A.; Osipova, E.S.; Safronov, S.V.; Epstein, L.M.; Shubina, E.S. Influence of phosphine (pincer) ligands on the transition metal hydrides reactivity. Coord. Chem. Rev. 2021, 438, 213799. [Google Scholar] [CrossRef]
- Slootweg, J.C. Sustainable Phosphorus Chemistry: A Silylphosphide Synthon for the Generation of Value-Added Phosphorus Chemicals. Angew. Chem. Int. Ed. 2018, 57, 6386–6388. [Google Scholar] [CrossRef]
- Falk Øgaard, A.; Brod, E. Efficient Phosphorus Cycling in Food Production: Predicting the Phosphorus Fertilization Effect of Sludge from Chemical Wastewater Treatment. J. Agric. Food Chem. 2016, 64, 4821–4829. [Google Scholar] [CrossRef]
- Glueck, D.S. Topics in Current Chemistry: New Aspects in Phosphorus Chemistry III; Majoral, J.P., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; Volume 229, p. 121. [Google Scholar]
- Wani, A.A.; Carballo, J.J.G.; Jayaprakash, H.; Wörle, M.; Widera, A.; Togni, A.; Grützmacher, H. A Simple Manganese(I) Catalyst for the Efficient and Selective Hydrophosphination of Olefins with PH3, Primary, and Secondary Phosphanes. Chem. Eur. J. 2024, 30, e202303848. [Google Scholar] [CrossRef]
- Longmire, J.M.; Zhang, X. Synthesis of chiral phosphine ligands with aromatic backbones and their applications in asymmetric catalysis. Tetrahedron Lett. 1997, 38, 1725–1728. [Google Scholar] [CrossRef]
- Feng, J.-J.; Chen, X.-F.; Shi, M.; Duan, W.-L. Palladium-Catalyzed Asymmetric Addition of Diarylphosphines to Enones toward the Synthesis of Chiral Phosphines. J. Am. Chem. Soc. 2010, 132, 5562–5563. [Google Scholar] [CrossRef]
- Ding, B.; Zhang, Z.; Xu, Y.; Liu, Y.; Sugiya, M.; Imamoto, T.; Zhang, W. P-Stereogenic PCP Pincer–Pd Complexes: Synthesis and Application in Asymmetric Addition of Diarylphosphines to Nitroalkenes. Org. Lett. 2013, 15, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, Z.; Ding, B.; Liu, D.; Liu, Y.; Sugiya, M.; Imamoto, T.; Zhang, W. Asymmetric Michael addition of diphenylphosphine to β,γ-unsaturated α-keto esters catalyzed by a P-stereogenic pincer-Pd complex. Tetrahedron 2015, 71, 6832–6839. [Google Scholar] [CrossRef]
- Yang, X.-Y.; Gan, J.H.; Li, Y.; Pullarkat, S.A.; Leung, P.-H. Palladium catalyzed asymmetric hydrophosphination of α,β- and α,β,γ,δ-unsaturated malonate esters—Efficient control of reactivity, stereo- and regio-selectivity. Dalton Trans. 2015, 44, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-J.; Liu, Y.-J.; Gong, J.-F.; Song, M.-P. Unsymmetrical Chiral PCN Pincer Palladium(II) and Nickel(II) Complexes with Aryl-Based Aminophosphine–Imidazoline Ligands: Synthesis via Aryl C–H Activation and Asymmetric Addition of Diarylphosphines to Enones. Organometallics 2011, 30, 3793–3803. [Google Scholar] [CrossRef]
- Hao, X.-Q.; Huang, J.-J.; Wang, T.; Lv, J.; Gong, J.-F.; Song, M.-P. PCN Pincer Palladium(II) Complex Catalyzed Enantioselective Hydrophosphination of Enones: Synthesis of Pyridine-Functionalized Chiral Phosphine Oxides as NCsp3O Pincer Preligands. J. Org. Chem. 2014, 79, 9512–9530. [Google Scholar] [CrossRef]
- Wang, C.; Huang, K.; Ye, J.; Duan, W.-L. Asymmetric Synthesis of P-Stereogenic Secondary Phosphine-Boranes by an Unsymmetric Bisphosphine Pincer-Nickel Complex. J. Am. Chem. Soc. 2021, 143, 5685–5690. [Google Scholar] [CrossRef]
- Wang, C.; Yin, P.; Dai, Y.-H.; Ye, J.; Duan, W.-L. Pincer-nickel catalyzed asymmetric addition of HPPh2 to enones toward the synthesis of chiral phosphines. J. Organomet. Chem. 2023, 983, 122552. [Google Scholar] [CrossRef]
- Wang, C.; Yang, Q.; Dai, Y.-H.; Xiong, J.; Zheng, Y.; Duan, W.-L. Nickel-Catalyzed Asymmetric Synthesis of P-Stereogenic Phosphanyl Hydrazine Building Blocks. Angew. Chem. Int. Ed. 2023, 62, e202313112. [Google Scholar] [CrossRef]
- Huang, J.-J.; Zhang, X.-Q.; Yang, J.-J.; Gong, J.-F.; Song, M.-P. Chiral (phosphine)-(imidazoline) PCN pincer palladium(ii) complexes: Synthesis and application in asymmetric hydrophosphination of 2-alkenoylpyridines with diphenylphosphine. Dalton Trans. 2022, 51, 8350–8367. [Google Scholar] [CrossRef] [PubMed]
- Pérez, J.M.; Postolache, R.; Castiñeira Reis, M.; Sinnema, E.G.; Vargová, D.; de Vries, F.; Otten, E.; Ge, L.; Harutyunyan, S.R. Manganese(I)-Catalyzed H–P Bond Activation via Metal–Ligand Cooperation. J. Am. Chem. Soc. 2021, 143, 20071–20076. [Google Scholar] [CrossRef]
- Smirnov, P.; Katan, E.; Mathew, J.; Kostenko, A.; Karni, M.; Nijs, A.; Bolm, C.; Apeloig, Y.; Marek, I. Formation of Three New Bonds and Two Stereocenters in Acyclic Systems by Zinc-Mediated Enantioselective Alkynylation of Acylsilanes, Brook Rearrangement, and Ene-Allene Carbocyclization Reactions. J. Org. Chem. 2014, 79, 12122–12135. [Google Scholar] [CrossRef] [PubMed]
- Kotani, S.; Kukita, K.; Tanaka, K.; Ichibakase, T.; Nakajima, M. Lithium Binaphtholate-Catalyzed Asymmetric Addition of Lithium Acetylides to Carbonyl Compounds. J. Org. Chem. 2014, 79, 4817–4825. [Google Scholar] [CrossRef]
- Smirnov, P.; Mathew, J.; Nijs, A.; Katan, E.; Karni, M.; Bolm, C.; Apeloig, Y.; Marek, I. One-Pot Zinc-Promoted Asymmetric Alkynylation/Brook-Type Rearrangement/Ene–Allene Cyclization: Highly Selective Formation of Three New Bonds and Two Stereocenters in Acyclic Systems. Angew. Chem. Int. Ed. 2013, 52, 13717–13721. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, R.; Kang, Y.-F.; Chen, C.; Xu, Z.-Q.; Zhou, Y.-F.; Ni, M.; Cai, H.-Q.; Gong, M.-Z. Highly Enantioselective Phenylacetylene Addition to Aromatic Ketones Catalyzed by Cinchona Alkaloid−Aluminum Complexes. J. Org. Chem. 2005, 70, 1084–1086. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kukita, K.; Ichibakase, T.; Kotani, S.; Nakajima, M. Lithium acetylides as alkynylating reagents for the enantioselective alkynylation of ketones catalyzed by lithium binaphtholate. Chem. Commun. 2011, 47, 5614–5616. [Google Scholar] [CrossRef]
- Cook, A.M.; Wolf, C. Efficient Access to Multifunctional Trifluoromethyl Alcohols through Base-Free Catalytic Asymmetric C−C Bond Formation with Terminal Ynamides. Angew. Chem. Int. Ed. 2016, 55, 2929–2933. [Google Scholar] [CrossRef]
- Chen, Q.; Tang, Y.; Huang, T.; Liu, X.; Lin, L.; Feng, X. Copper/Guanidine-Catalyzed Asymmetric Alkynylation of Isatins. Angew. Chem. Int. Ed. 2016, 55, 5286–5289. [Google Scholar] [CrossRef]
- Ohshima, T.; Kawabata, T.; Takeuchi, Y.; Kakinuma, T.; Iwasaki, T.; Yonezawa, T.; Murakami, H.; Nishiyama, H.; Mashima, K. C1-Symmetric Rh/Phebox-Catalyzed Asymmetric Alkynylation of α-Ketoesters. Angew. Chem. Int. Ed. 2011, 50, 6296–6300. [Google Scholar] [CrossRef]
- Morisaki, K.; Sawa, M.; Nomaguchi, J.-y.; Morimoto, H.; Takeuchi, Y.; Mashima, K.; Ohshima, T. Rh-Catalyzed Direct Enantioselective Alkynylation of α-Ketiminoesters. Chem. Eur. J. 2013, 19, 8417–8420. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.-I.; Ubukata, S.; Muraoka, S.; Nishiyama, H. Enantioselective Direct Alkynylation of Ketones Catalyzed by Chiral CCN Pincer RhIII Complexes. Chem. Eur. J. 2016, 22, 16801–16804. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, C.-Y.; Liu, J.-K.; Song, M.-P.; Gong, J.-F. Rhodium-Catalyzed Direct Enantioselective Alkynylation of Trifluoropyruvates with Terminal 1,3-Diynes. Adv. Synth. Catal. 2023, 365, 3967–3972. [Google Scholar] [CrossRef]
- Wang, Y.-D.; Liu, J.-K.; Yang, X.-J.; Gong, J.-F.; Song, M.-P. Chiral (Pyridine)-(Imidazoline) NCN′ Pincer Palladium(II) Complexes: Convenient Synthesis via C–H Activation and Characterization. Organometallics 2022, 41, 984–996. [Google Scholar] [CrossRef]
- Li, N.; Yang, X.; Zhu, Y.; Wang, F.; Gong, J.; Song, M. Pincer iridium(III)-catalyzed enantioselective C(sp3)-H functionalization via carbenoid CH insertion of 3-diazooxindoles with 1,4-cyclohexadiene. Chin. Chem. Lett. 2022, 33, 2437–2441. [Google Scholar] [CrossRef]
- Yokota, T.; Masu, H.; Arai, T. Asymmetric Friedel–Crafts-Type Reaction of 2-Vinylindoles to N-Boc Imines Using a Chiral Imidazolidine-Containing NCN-Pincer Pd Catalyst. J. Org. Chem. 2023, 88, 7872–7881. [Google Scholar] [CrossRef]
- Naganawa, Y.; Kawagishi, M.; Ito, J.-I.; Nishiyama, H. Asymmetric Induction at Remote Quaternary Centers of Cyclohexadienones by Rhodium-Catalyzed Conjugate Hydrosilylation. Angew. Chem. Int. Ed. 2016, 55, 6873–6876. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, Y.; Tsuchiya, Y.; Kobayashi, K.; Shiomi, T.; Itoh, J.-I.; Kikuchi, M.; Yamamoto, Y.; Nishiyama, H. Asymmetric Conjugate Reduction of α,β-Unsaturated Ketones and Esters with Chiral Rhodium(2,6-bisoxazolinylphenyl) Catalysts. Chem. Eur. J. 2006, 12, 63–71. [Google Scholar] [CrossRef]
- Nishiyama, H.; Shiomi, T.; Tsuchiya, Y.; Matsuda, I. High Performance of Rh(Phebox) Catalysts in Asymmetric Reductive Aldol Reaction: High Anti-Selectivity. J. Am. Chem. Soc. 2005, 127, 6972–6973. [Google Scholar] [CrossRef]
- Stathakis, C.I.; Gkizis, P.L.; Zografos, A.L. Metal-catalyzed cycloisomerization as a powerful tool in the synthesis of complex sesquiterpenoids. Nat. Prod. Rep. 2016, 33, 1093–1117. [Google Scholar] [CrossRef]
- Aubert, C.; Buisine, O.; Malacria, M. The Behavior of 1,n-Enynes in the Presence of Transition Metals. Chem. Rev. 2002, 102, 813–834. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-X.; Xu, S.-Q.; Han, Y.-P.; Liang, Y.-M. Recent Advances in Cyclization Reactions of 1,6-Enynes. Adv. Synth. Catal. 2024, 366, 1220–1268. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Wang, F.; Yang, D.-D.; Kittakoop, P.; Tan, Y.-X.; Tian, P. Enantioselective Reductive Cyclization of Alkynyl-Tethered Cyclohexadienones Catalyzed by Rhodium Complexes. Org. Lett. 2024, 26, 5614–5619. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Yu, H.-Y.; Xu, H.; Wang, Y.-J.; Yang, X.; Wang, Y.-H.; Tian, P.; Lin, G.-Q. Rhodium(III)-catalyzed diastereo- and enantioselective hydrosilylation/cyclization reaction of cyclohexadienone-tethered α,β-unsaturated aldehydes. Chin. Chem. Lett. 2024, 35, 109520. [Google Scholar] [CrossRef]
- Nishiyama, H.; Ito, J.-I. Bis(oxazolinyl)phenyl transition-metal complexes: Asymmetric catalysis and some reactions of the metals. Chem. Commun. 2010, 46, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Shu, T.; Cossy, J. Asymmetric desymmetrization of alkene-, alkyne- and allene-tethered cyclohexadienones using transition metal catalysis. Chem. Soc. Rev. 2021, 50, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Yoneda, T.; Ito, J.-I.; Nishiyama, H. Enantioselective Hydrosilylation of Aromatic Alkenes Catalyzed by Chiral Bis(oxazolinyl)phenyl–Rhodium Acetate Complexes. Synlett 2012, 23, 2957–2960. [Google Scholar] [CrossRef]
- Sangtrirutnugul, P.; Tilley, T.D. Silyl Derivatives of [Bis(8-quinolyl)methylsilyl]iridium(III) Complexes: Catalytic Redistribution of Arylsilanes and Dehydrogenative Arene Silylation. Organometallics 2007, 26, 5557–5568. [Google Scholar] [CrossRef]
- Kitano, T.; Komuro, T.; Ono, R.; Tobita, H. Tandem Hydrosilylation/o-C–H Silylation of Arylalkynes Catalyzed by Ruthenium Bis(silyl) Aminophosphine Complexes. Organometallics 2017, 36, 2710–2713. [Google Scholar] [CrossRef]
- Yang, B.; Gao, J.; Tan, X.; Ge, Y.; He, C. Chiral PSiSi-Ligand Enabled Iridium-Catalyzed Atroposelective Intermolecular C−H Silylation. Angew. Chem. Int. Ed. 2023, 62, e202307812. [Google Scholar] [CrossRef]
- Toribatake, K.; Zhou, L.; Tsuruta, A.; Nishiyama, H. Asymmetric β-boration of α,β-unsaturated carbonyl compounds with chiral Rh[bis(oxazolinyl)phenyl] catalysts. Tetrahedron 2013, 69, 3551–3560. [Google Scholar] [CrossRef]
- Toribatake, K.; Nishiyama, H. Asymmetric Diboration of Terminal Alkenes with a Rhodium Catalyst and Subsequent Oxidation: Enantioselective Synthesis of Optically Active 1,2-Diols. Angew. Chem. Int. Ed. 2013, 52, 11011–11015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, C.; Zhao, W. Enantio- and Regioselective Dihydroboration of Enals. ACS Catal. 2024, 14, 3977–3985. [Google Scholar] [CrossRef]
- Musa, S.; Ghosh, A.; Vaccaro, L.; Ackermann, L.; Gelman, D. Efficient E-Selective Transfer Semihydrogenation of Alkynes by Means of Ligand-Metal Cooperating Ruthenium Catalyst. Adv. Synth. Catal. 2015, 357, 2351–2357. [Google Scholar] [CrossRef]
- Musa, S.; Shaposhnikov, I.; Cohen, S.; Gelman, D. Ligand–Metal Cooperation in PCP Pincer Complexes: Rational Design and Catalytic Activity in Acceptorless Dehydrogenation of Alcohols. Angew. Chem. Int. Ed. 2011, 50, 3533–3537. [Google Scholar] [CrossRef]
- Cohen, O.; Grossman, O.; Vaccaro, L.; Gelman, D. Synthesis of chiral nonracemic PC(sp3)P pincer ligands. J. Organomet. Chem. 2014, 750, 13–16. [Google Scholar] [CrossRef]
- Radchenko, Y.; Mujahed, S.; Musa, S.; Gelman, D. Synthesis and characterization of chiral enantiopure PC(sp3)P pincer ligands and their complexes. Inorganica Chim. Acta 2021, 521, 120350. [Google Scholar] [CrossRef]






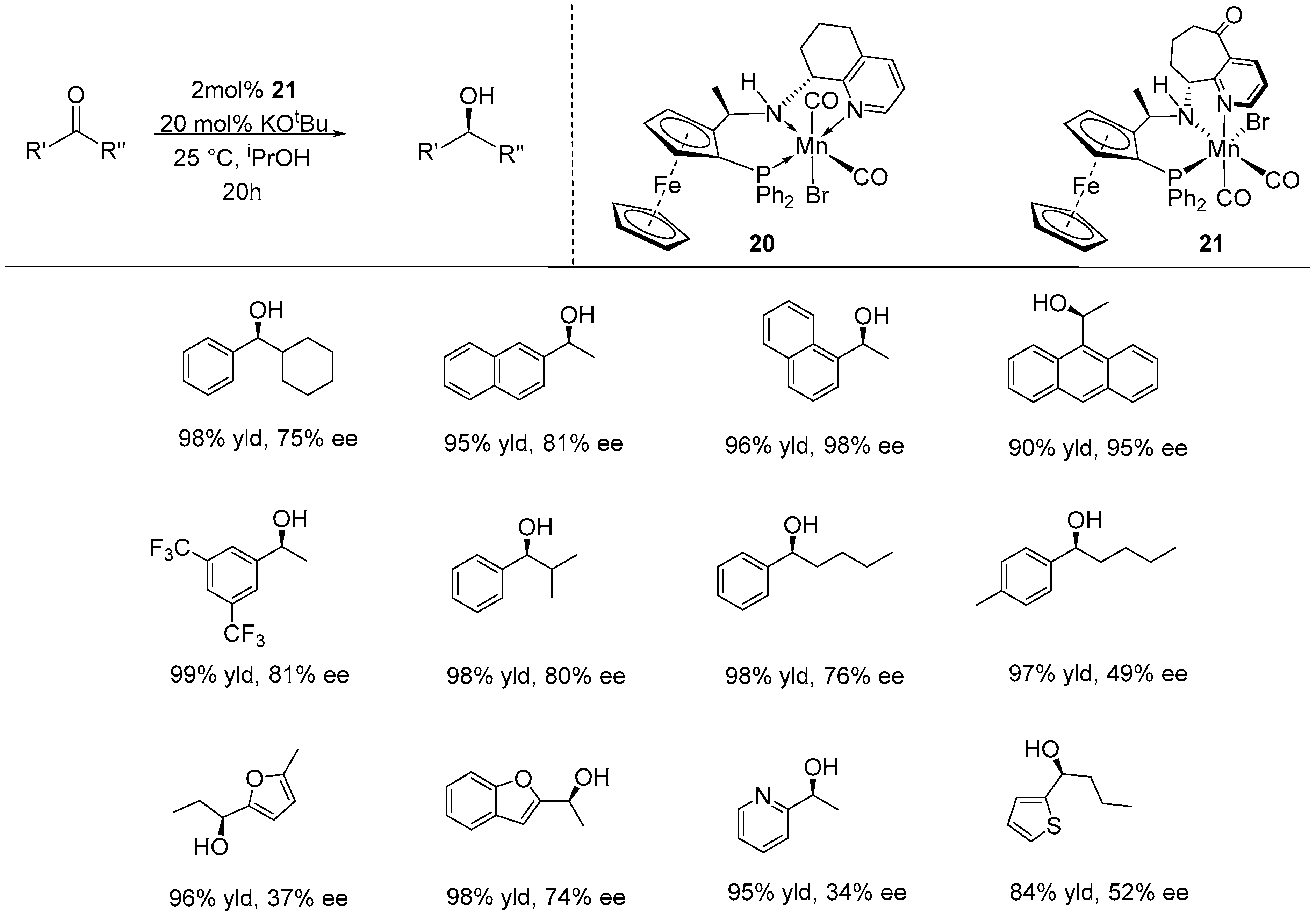

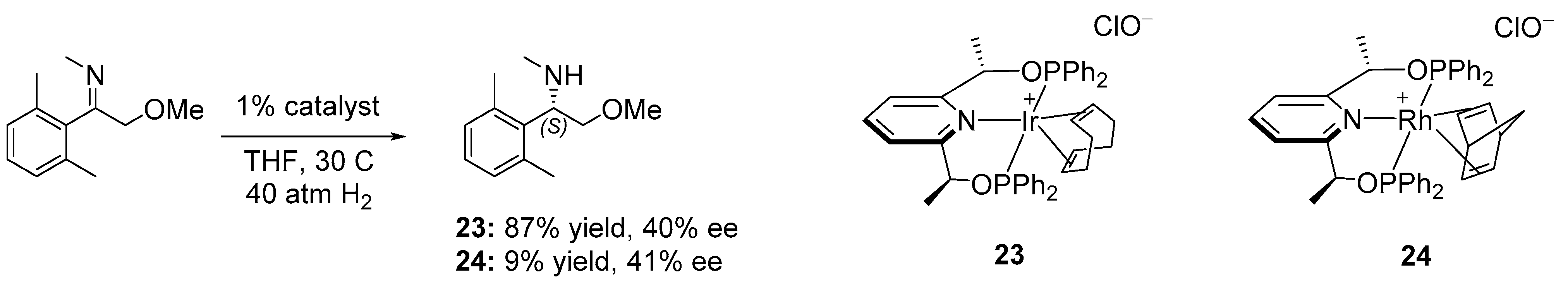
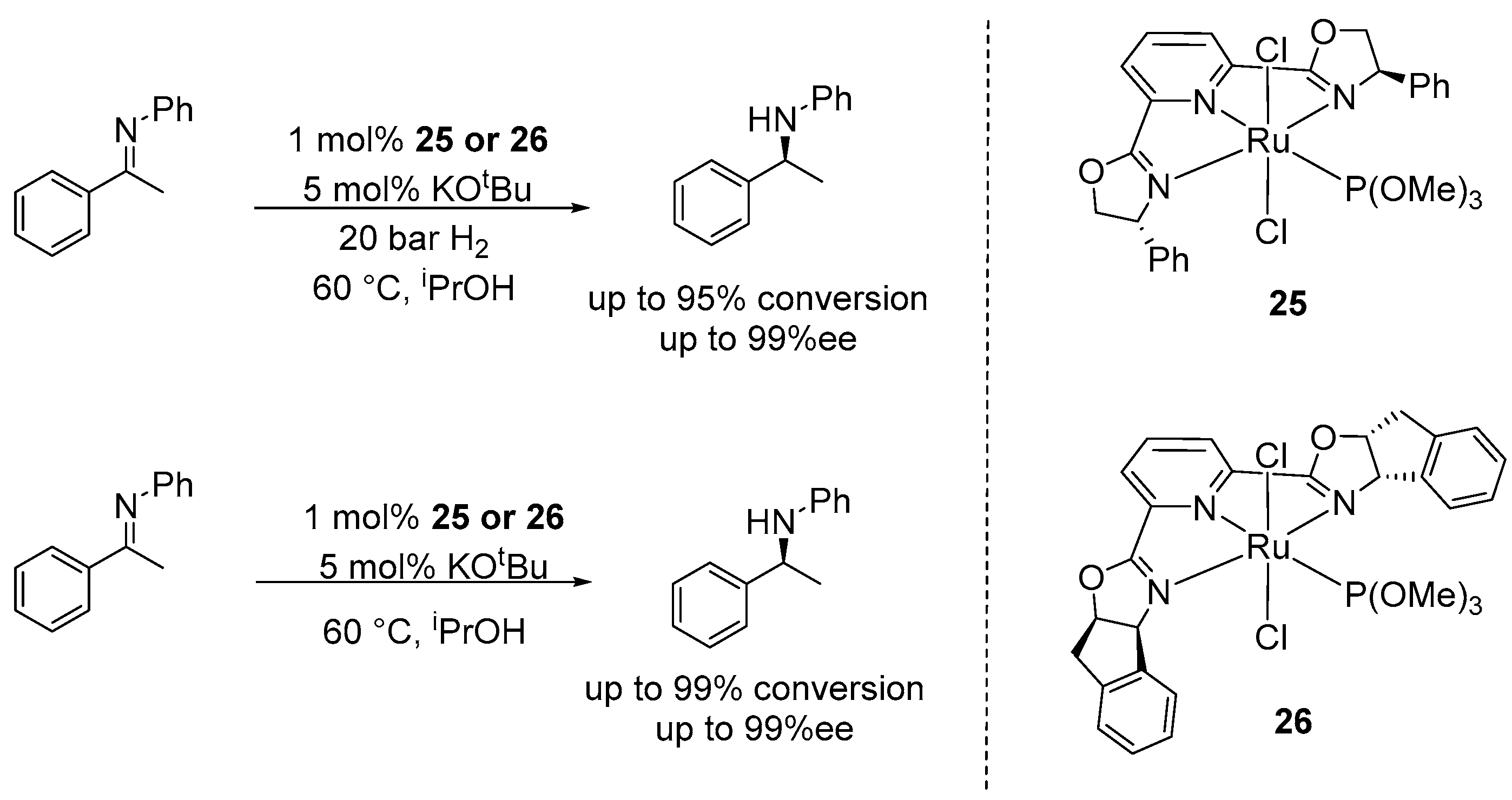
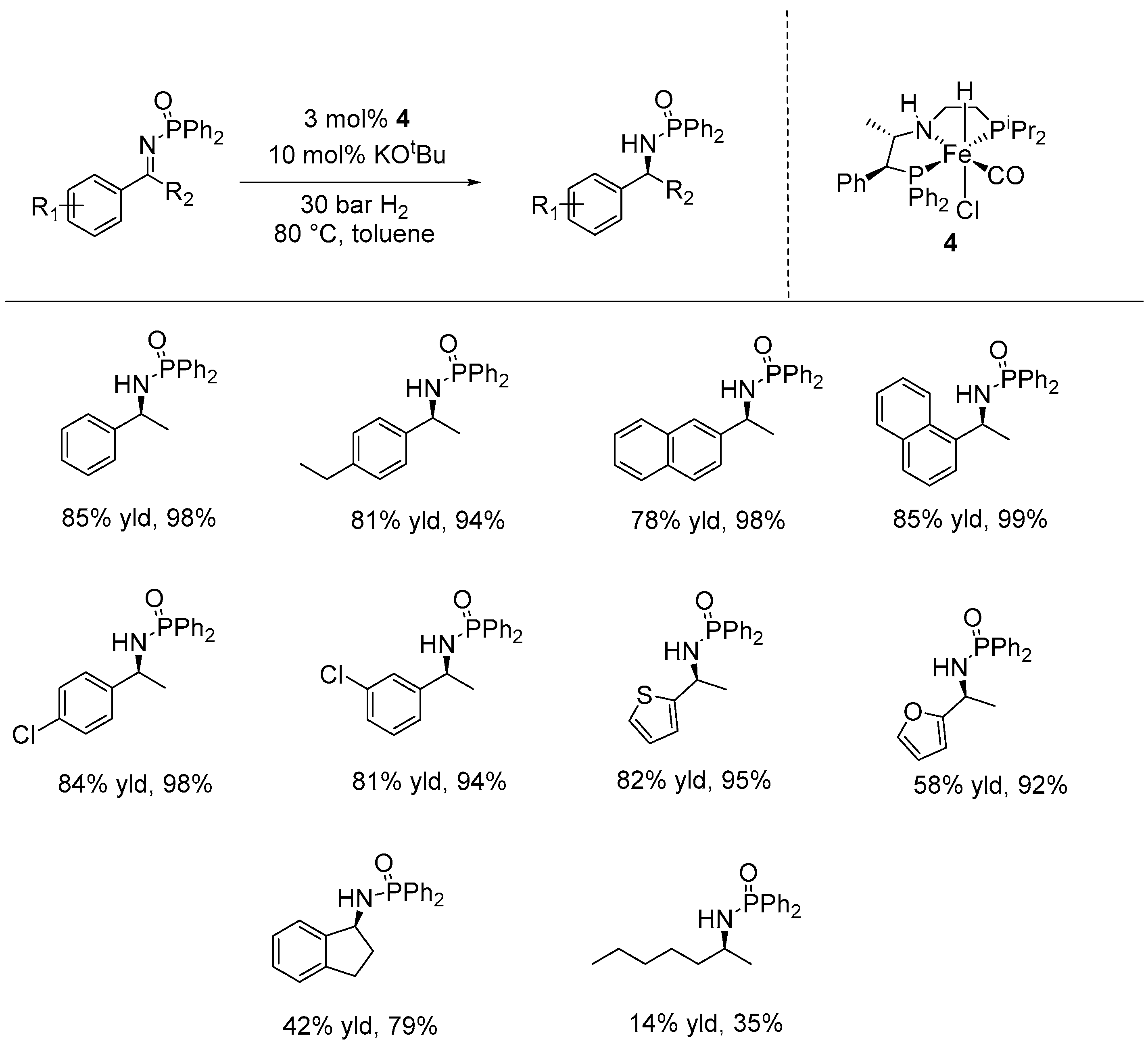






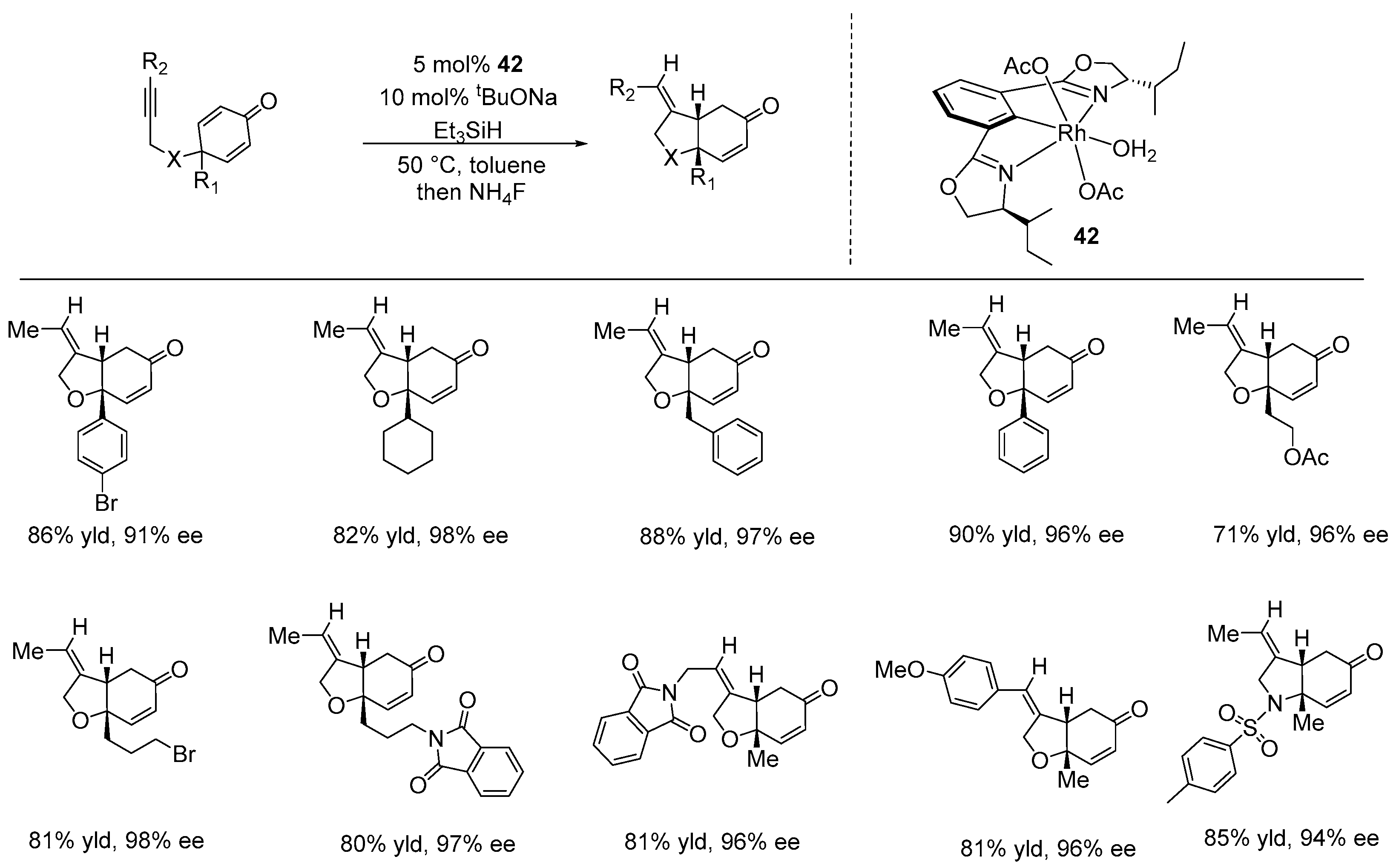

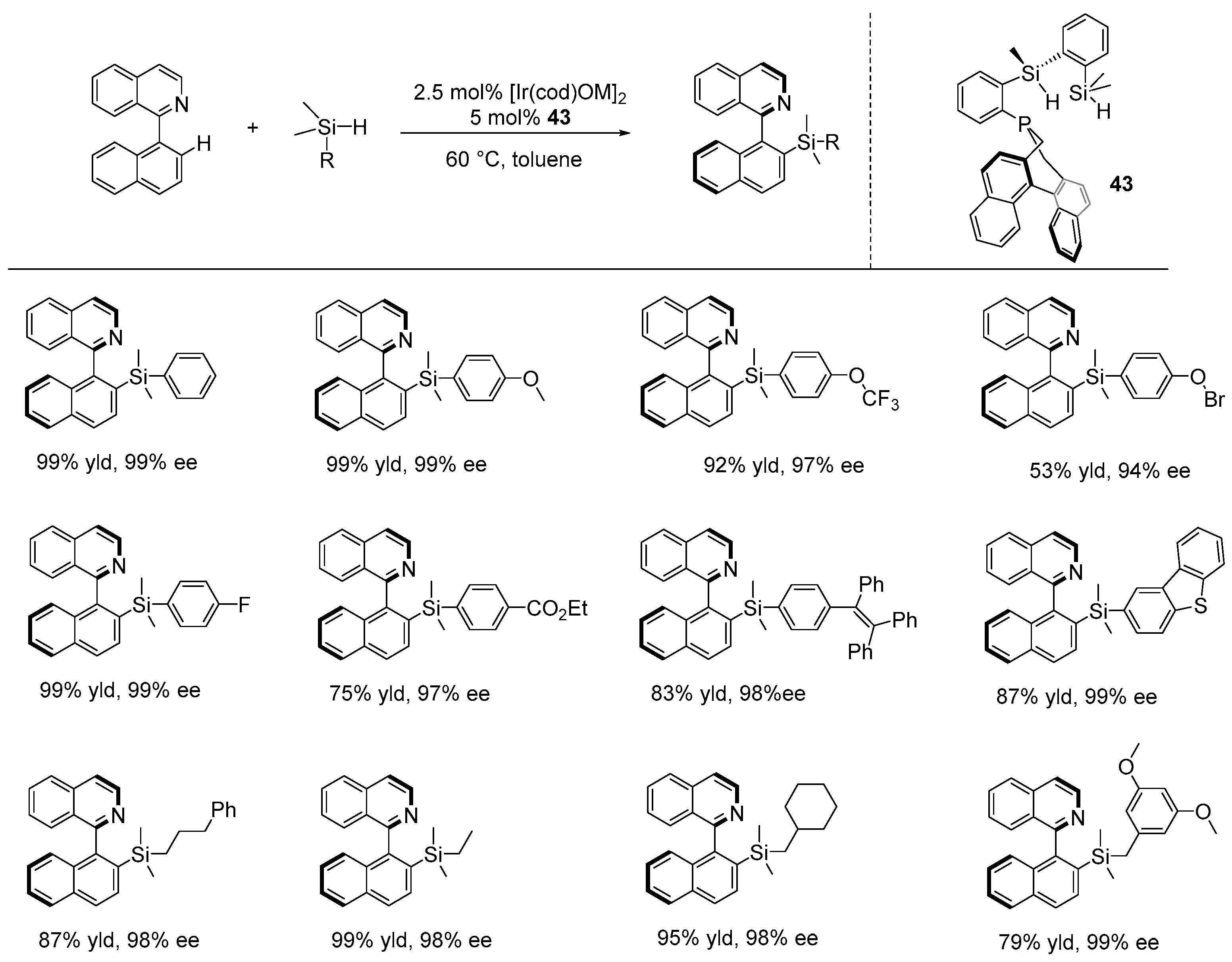
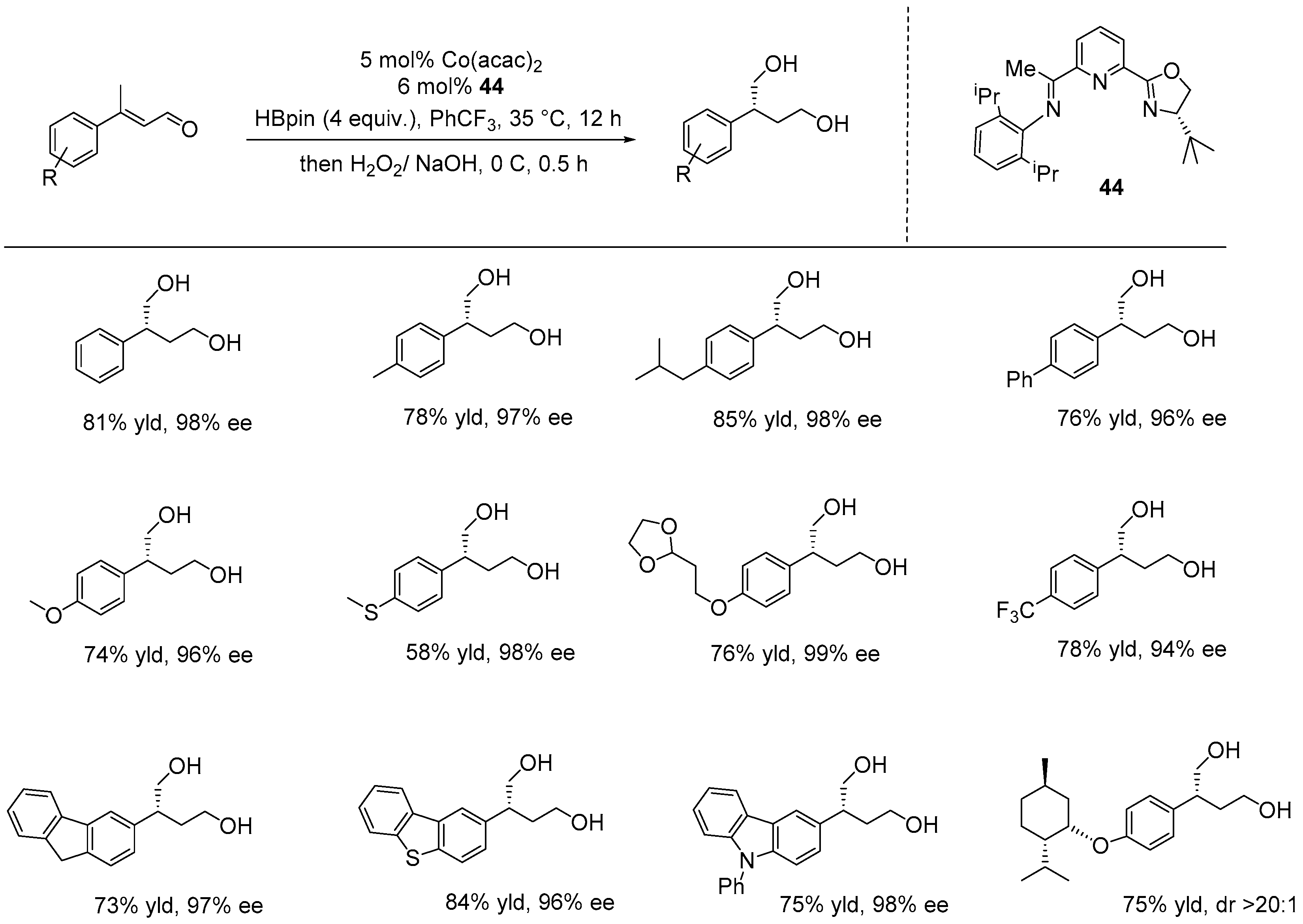

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musa, S.; Peretz, Y.; Dinnar, G. Advances in Chiral Pincer Complexes: Insights and Applications in Catalytic Asymmetric Reactions. Int. J. Mol. Sci. 2024, 25, 10344. https://doi.org/10.3390/ijms251910344
Musa S, Peretz Y, Dinnar G. Advances in Chiral Pincer Complexes: Insights and Applications in Catalytic Asymmetric Reactions. International Journal of Molecular Sciences. 2024; 25(19):10344. https://doi.org/10.3390/ijms251910344
Chicago/Turabian StyleMusa, Sanaa, Yuval Peretz, and Gil Dinnar. 2024. "Advances in Chiral Pincer Complexes: Insights and Applications in Catalytic Asymmetric Reactions" International Journal of Molecular Sciences 25, no. 19: 10344. https://doi.org/10.3390/ijms251910344
APA StyleMusa, S., Peretz, Y., & Dinnar, G. (2024). Advances in Chiral Pincer Complexes: Insights and Applications in Catalytic Asymmetric Reactions. International Journal of Molecular Sciences, 25(19), 10344. https://doi.org/10.3390/ijms251910344