
Effect of CFTR Modulators on Oxidative Stress and Autophagy in Non-CFTR-Expressing Cells

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
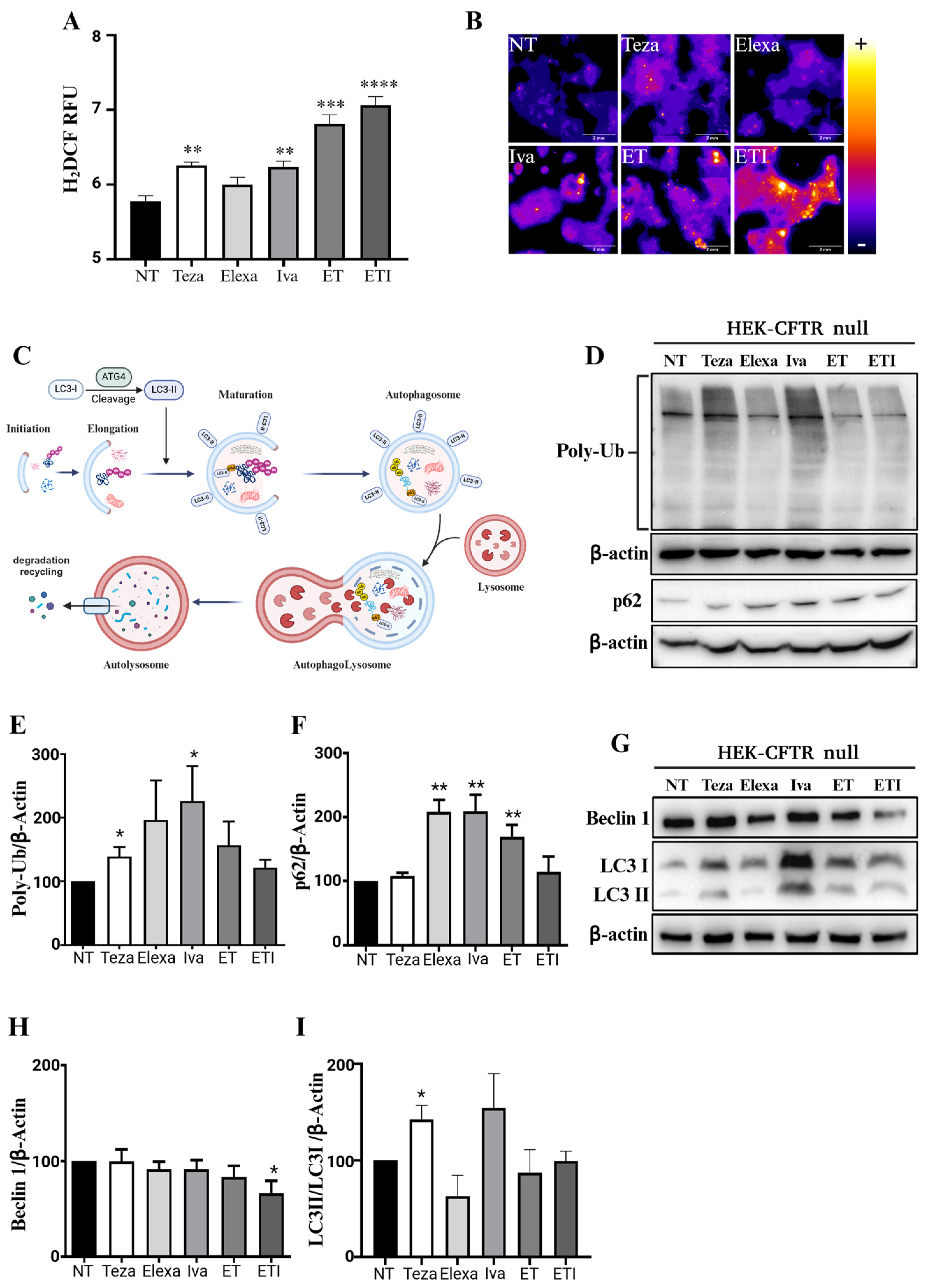
2.1. CFTR Modulators Increase Reactive Oxygen Species and Autophagic Markers in CFTR-Null HEK Cells
2.2. ET and ETI Modulate Rab5 and Rab7 in HEK-CFTR Null Cells
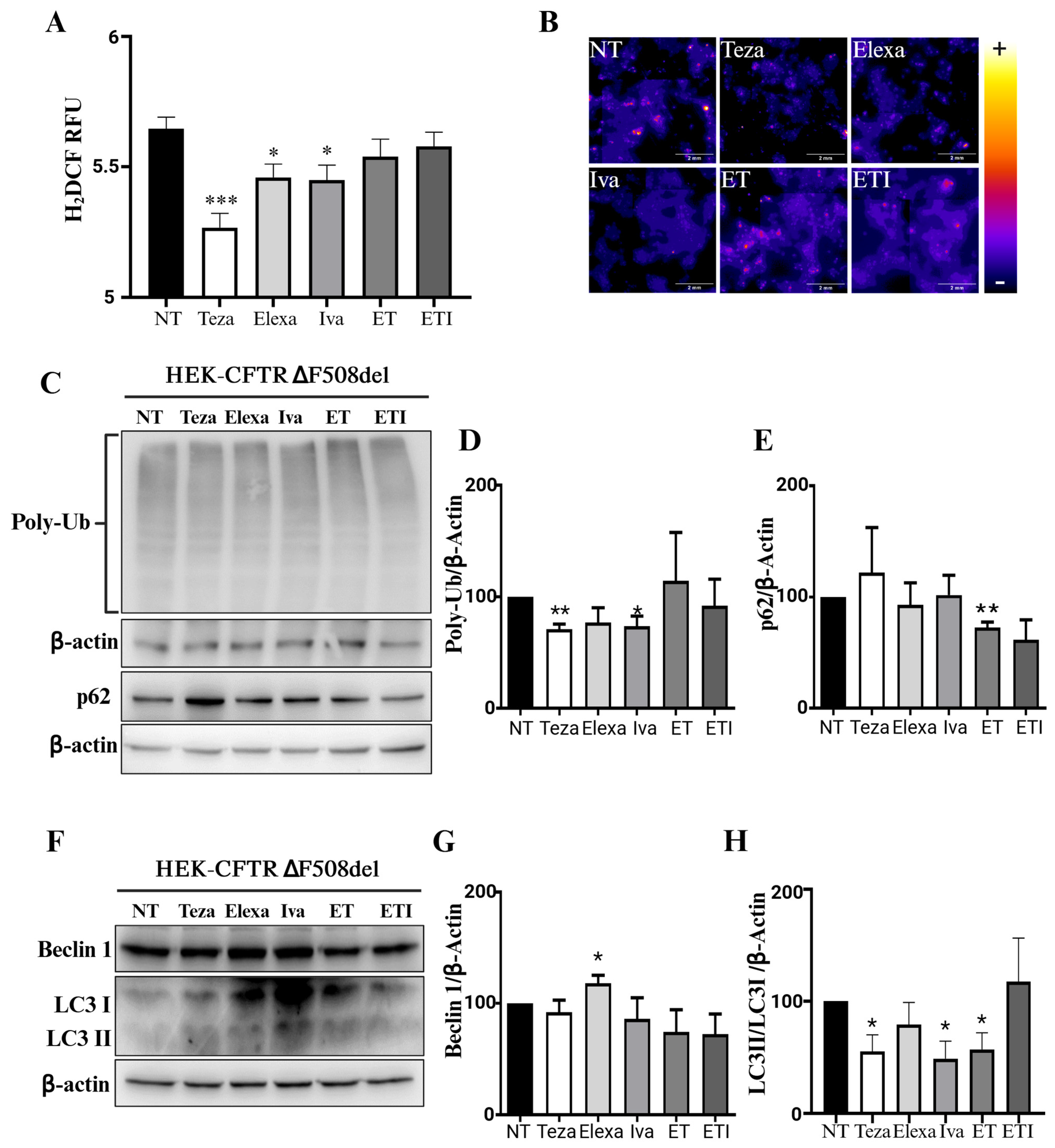
2.3. CFTR Modulators Do Not Affect Autophagy in CFTR-∆F508del HEK Cells
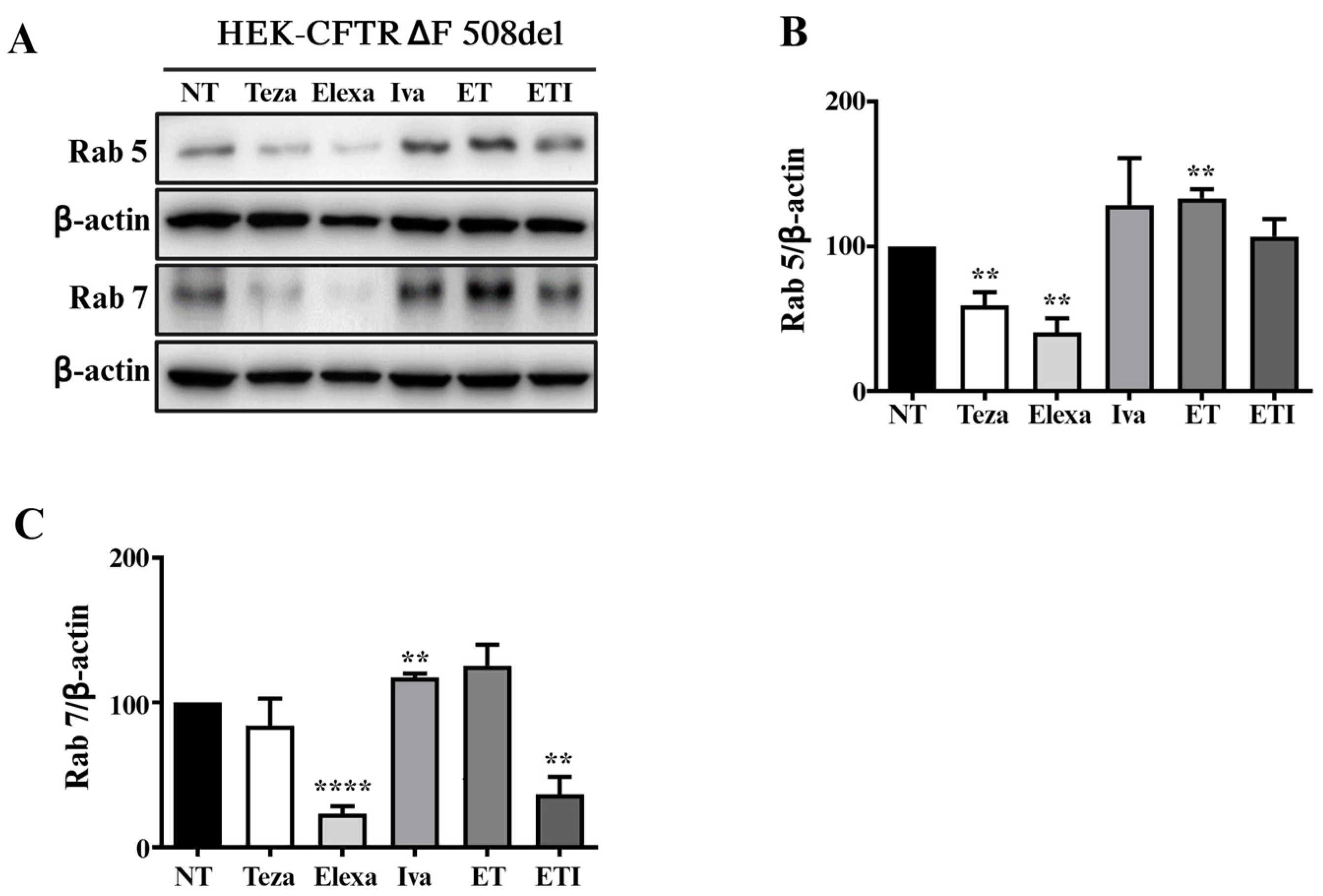
2.4. Rab5 and Rab7 Are Differently Regulated in HEK-CFTR-∆F508del Cells Treated with CFTR Modulators
3. Materials and Methods
3.1. Cell Culture and Generation of HEK293 Cells Expressing the F508del-CFTR
3.2. Cell Treatments
3.3. Western Blot
3.4. Reactive Oxygen Species Detection
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef] [PubMed]
- Accurso, F.J. Update in cystic fibrosis 2005. Am. J. Respir. Crit. Care Med. 2006, 173, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Nam, J.H.; Kim, J.Y.; Namkung, W.; Yoon, J.S.; Lee, J.S.; Kim, K.S.; Venglovecz, V.; Gray, M.A.; Kim, K.H.; et al. Dynamic regulation of CFTR bicarbonate permeability by [Cl−]i and its role in pancreatic bicarbonate secretion. Gastroenterology 2010, 139, 620–631. [Google Scholar] [CrossRef]
- Quinton, P.M. Physiological basis of cystic fibrosis: A historical perspective. Physiol. Rev. 1999, 79 (Suppl. S1), S3–S22. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S. Cystic fibrosis: Molecular biology and therapeutic implications. Science 1992, 256, 774–779. [Google Scholar] [CrossRef]
- Durupt, S.; Mazur, S.; Reix, P. Therapeutic advances in cystic fibrosis in 2014. Rev. Pneumol. Clin. 2016, 72, 77–86. [Google Scholar] [CrossRef]
- This Patient Information Has Been Approved by the U.S. Food and Drug Administration. Revised: 08/2023. Available online: https://pi.vrtx.com/files/uspi_elexacaftor_tezacaftor_ivacaftor.pdf (accessed on 23 September 2024).
- Dreano, E.; Burgel, P.R.; Hatton, A.; Bouazza, N.; Chevalier, B.; Macey, J.; Leroy, S.; Durieu, I.; Weiss, L.; Grenet, D.; et al. Theratyping cystic fibrosis patients to guide elexacaftor/tezacaftor/ivacaftor out-of-label prescription. Eur. Respir. J. 2023, 62, 2300110. [Google Scholar] [CrossRef]
- Ridley, K.; Condren, M. Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy. J. Pediatr. Pharmacol. Ther. 2020, 25, 192–197. [Google Scholar] [CrossRef]
- He, R.; Lin, F.; Deng, Z.; Yu, B. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with Phe508del mutation: Evidence from randomized controlled trials. SAGE Open Med. 2024, 12, 20503121231225874. [Google Scholar] [CrossRef]
- Burgel, P.R.; Paillasseur, J.L.; Durieu, I.; Reynaud-Gaubert, M.; Hamidfar, R.; Murris-Espin, M.; Danner-Boucher, I.; Chiron, R.; Leroy, S.; Douvry, B.; et al. Multisystemic Effects of Elexacaftor-Tezacaftor-Ivacaftor in Adults with Cystic Fibrosis and Advanced Lung Disease. Ann. Am. Thorac. Soc. 2024, 21, 1053–1064. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Robinson, P.D.; Shteinberg, M.; Downey, D.G. CFTR modulator therapy: Transforming the landscape of clinical care in cystic fibrosis. Lancet 2023, 402, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- A Phase 3 Study of VX-445 Combination Therapy in Subjects with Cystic Fibrosis Heterozygous for the F508del Mutation and a Minimal Function Mutation (F/MF). 2020; Last Update Posted 2020-05-19. Available online: https://clinicaltrials.gov/ct2/show/NCT03525444 (accessed on 23 September 2024).
- Castaldo, A.; Iacotucci, P.; Bagnasco, S.; Fevola, C.; Carnovale, V.; Antonelli, F.; Cernera, G.; Gelzo, M.; Terlizzi, V. Liver biochemical indexes and cholesterol metabolism in cystic fibrosis patients with F508del/CFTR variant genotype after elexacaftor/tezacaftor/ivacaftor treatment. Sci. Rep. 2024, 14, 17422. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, A.; Gelzo, M.; Iacotucci, P.; Longobardi, A.; Taccetti, G.; Terlizzi, V.; Carnovale, V. One year of treatment with elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis homozygous for the F508del mutation causes a significant increase in liver biochemical indexes. Front. Mol. Biosci. 2024, 10, 1327958. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Fevola, C.; Presti, S.; Castaldo, A.; Daccò, V.; Claut, L.; Sepe, A.; Majo, F.; Casciaro, R.; Esposito, I.; et al. Reported Adverse Events in a Multicenter Cohort of Patients Ages 6–18 Years with Cystic Fibrosis and at Least One F508del Allele Receiving Elexacaftor/Tezacaftor/Ivacaftor. J. Pediatr. 2024, 274, 114176. [Google Scholar] [CrossRef]
- Carnovale, V.; Scialò, F.; Gelzo, M.; Iacotucci, P.; Amato, F.; Zarrilli, F.; Celardo, A.; Castaldo, G.; Corso, G. Cystic Fibrosis Patients with F508del/Minimal Function Genotype: Laboratory and Nutritional Evaluations after One Year of Elexacaftor/Tezacaftor/Ivacaftor Treatment. J. Clin. Med. 2022, 11, 6900. [Google Scholar] [CrossRef]
- Piona, C.; Mozzillo, E.; Tosco, A.; Volpi, S.; Rosanio, F.M.; Cimbalo, C.; Franzese, A.; Raia, V.; Zusi, C.; Emiliani, F.; et al. Impact of CFTR Modulators on Beta-Cell Function in Children and Young Adults with Cystic Fibrosis. J. Clin. Med. 2022, 11, 4149. [Google Scholar] [CrossRef]
- Ciobanu, D.Z.; Liessi, N.; Tomati, V.; Capurro, V.; Bertozzi, S.M.; Summa, M.; Bertorelli, R.; Loberto, N.; Dobi, D.; Aureli, M.; et al. Tezacaftor is a direct inhibitor of sphingolipid delta-4 desaturase enzyme (DEGS). J. Cyst. Fibros. 2024; in press. [Google Scholar] [CrossRef]
- Liessi, N.; Tomati, V.; Capurro, V.; Loberto, N.; Garcia-Aloy, M.; Franceschi, P.; Aureli, M.; Pedemonte, N.; Armirotti, A. The combination elexacaftor/tezacaftor/ivacaftor (ETI) modulates the de novo synthethic pathway of ceramides in a genotype-independent manner. J. Cyst. Fibros. 2023, 22, 680–682. [Google Scholar] [CrossRef]
- Schneider, E.K.; McQuade, R.M.; Carbone, V.C.; Reyes-Ortega, F.; Wilson, J.W.; Button, B.; Saito, A.; Poole, D.P.; Hoyer, D.; Li, J.; et al. The potentially beneficial central nervous system activity profile of ivacaftor and its metabolites. ERJ Open Res. 2018, 4, 00127-2017. [Google Scholar] [CrossRef] [PubMed]
- Cigana, C.; Giannella, R.; Colavolpe, A.; Alcalá-Franco, B.; Mancini, G.; Colombi, F.; Bigogno, C.; Bastrup, U.; Bertoni, G.; Bragonzi, A. Mutual Effects of Single and Combined CFTR Modulators and Bacterial Infection in Cystic Fibrosis. Microbiol. Spectr. 2023, 11, e0408322. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.Y.; Lim, D.J.; Mackey, C.; Skinner, D.; Zhang, S.; McCormick, J.; Woodworth, B.A. Ivacaftor, a Cystic Fibrosis Transmembrane Conductance Regulator Potentiator, Enhances Ciprofloxacin Activity against Pseudomonas aeruginosa. Am. J. Rhinol. Allergy 2019, 33, 129–136. [Google Scholar] [CrossRef]
- Esposito, S.; Zollo, I.; Villella, V.R.; Scialò, F.; Giordano, S.; Esposito, M.V.; Salemme, N.; Di Domenico, C.; Cernera, G.; Zarrilli, F.; et al. Identification of an ultra-rare Alu insertion in the CFTR gene: Pitfalls and challenges in genetic test interpretation. Clin. Chim. Acta 2024, 558, 118317. [Google Scholar] [CrossRef]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef]
- Badr, A.; Eltobgy, M.; Krause, K.; Hamilton, K.; Estfanous, S.; Daily, K.P.; Abu Khweek, A.; Hegazi, A.; Anne, M.N.K.; Carafice, C.; et al. CFTR Modulators Restore Acidification of Autophago-Lysosomes and Bacterial Clearance in Cystic Fibrosis Macrophages. Front. Cell. Infect. Microbiol. 2022, 12, 819554. [Google Scholar] [CrossRef]
- McDonald, E.F.; Kim, M.; Olson, J.A., 3rd; Meiler, J.; Plate, L. Proteostasis Landscapes of Selective versus Poorly Responsive CFTR Variants Reveals Structural Vulnerabilities to Correction. bioRxiv 2024, preprint. [Google Scholar] [CrossRef]
- Villella, V.R.; Esposito, S.; Bruscia, E.M.; Vicinanza, M.; Cenci, S.; Guido, S.; Pettoello-Mantovani, M.; Carnuccio, R.; De Matteis, M.A.; Luini, A.; et al. Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ. 2013, 20, 1101–1115. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Gavina, M.; Russo, I.; Silano, M.; Guido, S.; Pettoello-Mantovani, M.; Carnuccio, R.; Scholte, B.; et al. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012, 8, 1657–1672. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef]
- Villella, V.R.; Esposito, S.; Ferrari, E.; Monzani, R.; Tosco, A.; Rossin, F.; Castaldo, A.; Silano, M.; Marseglia, G.L.; Romani, L.; et al. Autophagy suppresses the pathogenic immune response to dietary antigens in cystic fibrosis. Cell Death Dis. 2019, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Farinha, C.M.; Matos, P. Rab GTPases regulate the trafficking of channels and transporters—A focus on cystic fibrosis. Small GTPases 2018, 9, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- de Almeida, A.J.P.O.; de Oliveira, J.C.P.L.; da Silva Pontes, L.V.; de Souza Júnior, J.F.; Gonçalves, T.A.F.; Dantas, S.H.; de Almeida Feitosa, M.S.; Silva, A.O.; de Medeiros, I.A. ROS: Basic Concepts, Sources, Cellular Signaling, and its Implications in Aging Pathways. Oxid. Med. Cell. Longev. 2022, 2022, 1225578. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022, 185, 158–168.e11. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Available online: www.trikafta.com/safety-side-effects (accessed on 23 September 2024).
- Rachel, M.; Galiniak, S.; Biesiadecki, M.; Gala-Błądzińska, A. Renal Function in Patients with Cystic Fibrosis: A Single-Center Study. Int. J. Environ. Res. Public Health 2022, 19, 5454. [Google Scholar] [CrossRef] [PubMed]
- Nazareth, D.; Walshaw, M. A review of renal disease in cystic fibrosis. J. Cyst. Fibros. 2013, 12, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Denimal, D.; Bergas, V.; Pais-de-Barros, J.P.; Simoneau, I.; Demizieux, L.; Passilly-Degrace, P.; Bouillet, B.; Petit, J.M.; Rouland, A.; Bataille, A.; et al. Liraglutide reduces plasma dihydroceramide levels in patients with type 2 diabetes. Cardiovasc. Diabetol. 2023, 22, 104. [Google Scholar] [CrossRef]
- Choi, R.H.; Tatum, S.M.; Symons, J.D.; Summers, S.A.; Holland, W.L. Ceramides and other sphingolipids as drivers of cardiovascular disease. Nat. Rev. Cardiol. 2021, 18, 701–711. [Google Scholar] [CrossRef]
- Zarini, S.; Brozinick, J.T.; Zemski Berry, K.A.; Garfield, A.; Perreault, L.; Kerege, A.; Bui, H.H.; Sanders, P.; Siddall, P.; Kuo, M.S.; et al. Serum dihydroceramides correlate with insulin sensitivity in humans and decrease insulin sensitivity in vitro. J. Lipid Res. 2022, 63, 100270. [Google Scholar] [CrossRef]
- Edsfeldt, A.; Dunér, P.; Ståhlman, M.; Mollet, I.G.; Asciutto, G.; Grufman, H.; Nitulescu, M.; Persson, A.F.; Fisher, R.M.; Melander, O.; et al. Sphingolipids Contribute to Human Atherosclerotic Plaque Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1132–1140. [Google Scholar] [CrossRef]
- Heo, S.; Young, D.C.; Safirstein, J.; Bourque, B.; Antell, M.H.; Diloreto, S.; Rotolo, S.M. Mental status changes during elexacaftor/tezacaftor/ivacaftor therapy. J. Cyst. Fibros. 2022, 21, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Spoletini, G.; Gillgrass, L.; Pollard, K.; Shaw, N.; Williams, E.; Etherington, C.; Clifton, I.J.; Peckham, D.G. Dose adjustments of Elexacaftor/Tezacaftor/Ivacaftor in response to mental health side effects in adults with cystic fibrosis. J. Cyst. Fibros. 2022, 21, 1061–1065. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scialò, F.; Cernera, G.; Polise, L.; Castaldo, G.; Amato, F.; Villella, V.R. Effect of CFTR Modulators on Oxidative Stress and Autophagy in Non-CFTR-Expressing Cells. Int. J. Mol. Sci. 2024, 25, 10360. https://doi.org/10.3390/ijms251910360
Scialò F, Cernera G, Polise L, Castaldo G, Amato F, Villella VR. Effect of CFTR Modulators on Oxidative Stress and Autophagy in Non-CFTR-Expressing Cells. International Journal of Molecular Sciences. 2024; 25(19):10360. https://doi.org/10.3390/ijms251910360
Chicago/Turabian StyleScialò, Filippo, Gustavo Cernera, Lorenza Polise, Giuseppe Castaldo, Felice Amato, and Valeria Rachela Villella. 2024. "Effect of CFTR Modulators on Oxidative Stress and Autophagy in Non-CFTR-Expressing Cells" International Journal of Molecular Sciences 25, no. 19: 10360. https://doi.org/10.3390/ijms251910360