
Macrophages in the Context of Muscle Regeneration and Duchenne Muscular Dystrophy

 ,
,
Abstract
1. Introduction
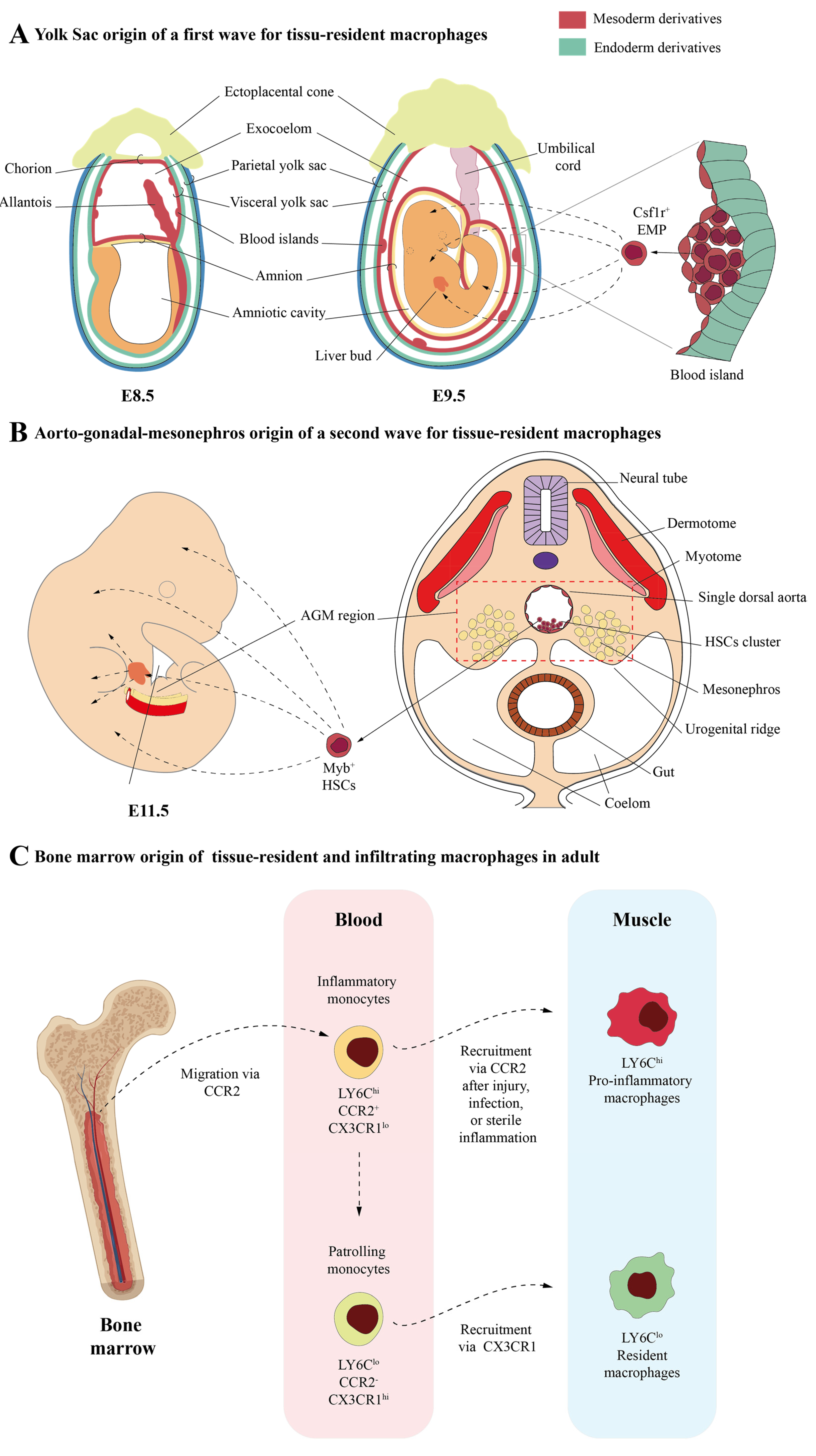
2. Macrophages in the Skeletal Muscle: Different Origins and Subtypes
2.1. Skeletal Muscle-Resident Macrophages
2.2. Infiltrating Macrophages
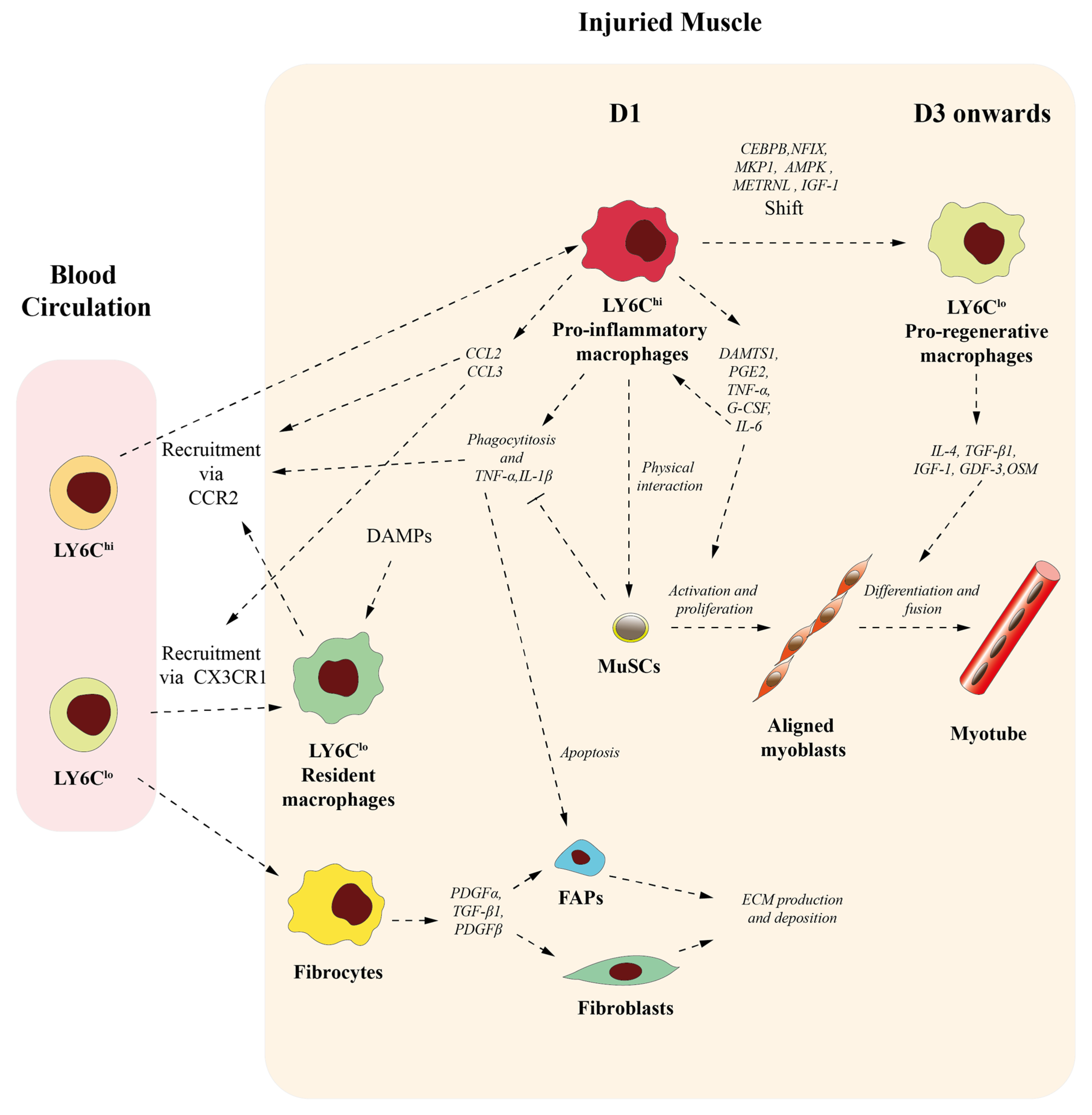
3. Roles of Macrophages on Skeletal Muscle Homeostasis and Muscle Repair
3.1. Macrophages Contribute to Initiate but Also Resolve Inflammation
3.2. Macrophages Participate in Muscle Stem Cell Activation and Differentiation
3.3. Macrophages Conditionate Muscle Repair-Microenvironment
4. Macrophages in DMD Microenvironment
5. Macrophages as Therapeutic Targets for Dystrophy DMD Treatment

{kind=link}
{kind=link}
| Molecular Target | Strategy/Effect | References |
|---|---|---|
| IL-10 | Treatment suppresses the production of pro-inflammatory cytokines. | Murray 2006; Mosser, Zhang 2008 [134,135] |
| Treatment facilitates the switch to the M2c phenotype. | Mantovani et al., 2004; Lang et al., 2002 [136,137] | |
| Treatment inhibits the expression of iNOS, thus enhancing muscle regeneration during DMD and preserving muscle function. | Villalta, Rinaldi, et al., 2011 Villalta, Deng, et al., 2011 [101,138] | |
| IFN-γ | Null mutation increases M2 phenotype activation during the regenerative stage in mdx [138]. | Villalta, Deng, et al., 2011 [138] |
| TNF | cV1q anti-TNF antibody improves muscle function, reduces myofiber leakiness, and reduces DMD severity. | Radley, et al., 2008 [142] |
| Infliximab anti-TNF antibody and/or Etanercept soluble TNF-receptor delays and reduces the breakdown of dystrophic muscle in young dystrophic mdx mice. | Grounds et al., 2004; Hodgetts et al., 2006 [143,144] | |
| TGF-β | Suramin inhibits the ability of TGF-β1 to bind to its receptors and promotes muscle regeneration, attenuates fibrosis in the diaphragm and limb muscles, and prevents exercise-induced functional muscle loss of mdx mice. | Chan et al., 2003; 2005; Nozaki et al., 2008; Coffey et al., 1987; La Rocca et al., 1990; McGeary et al., 2008;Taniguti et al., 2011 [145,146,147,148,149,150,151] |
| Metformin and/or activator 991 decreases TNF-α production, increases CD206 and CD30 production, decreases the necrotic and fibrotic areas, and increases cross-sectional area in mdx mice. | Foretz et al., 2014; Guigas et al., 2016; Juban et al., 2018 [108,152,153] | |
| CSF1R | PLX73086 ablates TIMD4+ and TIMD4− muscle-resident macrophages cells, thus shielding dystrophic muscles from eccentric contraction-induced injury. | Babaeijandaghi et al., 2022 [28] |
| NFIX | Deletion of Nfix in macrophages of dystrophic mice delays the establishment of fibrosis and muscle wasting and increases grasp force. | Saclier et al., 2022; Gronostajski 2000 [157,158] |
| Nfix silencing rescues dystrophic muscle morphology in terms of reduced infiltrates, centrally nucleated myofibers, and CSA distribution. | Rossi et al., 2017 [159] | |
| GSK3β | ILA decreases the expression of pro-inflammatory cytokines TNF-α, IL-1β, and MCP-1, diminishes muscle fibrosis, and boosts muscle regeneration in mdx. | Matias-Valiente et al., 2023 [167] |
| TLR4 | TLR4 ablation and/or Glycyrrhizin treatment decreases macrophage accumulation in mdx mice, also causing macrophages to acquire an anti-inflammatory phenotype. | Giordano et al., 2015 [112] |
6. Concluding Remarks and Future Research
Funding
Conflicts of Interest
Abbreviations
References
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage Biology in Development, Homeostasis and Disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Prinz, M.; Geissmann, F.; Gomez Perdiguero, E. Development and Function of Tissue Resident Macrophages in Mice. Semin. Immunol. 2015, 27, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Ginhoux, F.; Schultze, J.L.; Murray, P.J.; Ochando, J.; Biswas, S.K. New Insights into the Multidimensional Concept of Macrophage Ontogeny, Activation and Function. Nat. Immunol. 2016, 17, 34–40. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization, and Function in Health and Disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Tauber, A.I. Metchnikoff and the Phagocytosis Theory. Nat. Rev. Mol. Cell Biol. 2003, 4, 897–901. [Google Scholar] [CrossRef]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The Mononuclear Phagocyte System: A New Classification of Macrophages, Monocytes, and Their Precursor Cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191–2193. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory Monocytes Recruited after Skeletal Muscle Injury Switch into Antiinflammatory Macrophages to Support Myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Buscher, K.; Ehinger, E.; Gupta, P.; Pramod, A.B.; Wolf, D.; Tweet, G.; Pan, C.; Mills, C.D.; Lusis, A.J.; Ley, K. Natural Variation of Macrophage Activation as Disease-Relevant Phenotype Predictive of Inflammation and Cancer Survival. Nat. Commun. 2017, 8, 16041. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and Macrophage Differentiation: Circulation Inflammatory Monocyte as Biomarker for Inflammatory Diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A. Tissue Macrophages: Heterogeneity and Functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Sunderkötter, C.; Nikolic, T.; Dillon, M.J.; Van Rooijen, N.; Stehling, M.; Drevets, D.A.; Leenen, P.J.M. Subpopulations of Mouse Blood Monocytes Differ in Maturation Stage and Inflammatory Response. J. Immunol. 2004, 172, 4410–4417. [Google Scholar] [CrossRef] [PubMed]
- Hristov, M.; Weber, C. Differential Role of Monocyte Subsets in Atherosclerosis. Thromb. Haemost. 2011, 106, 757–762. [Google Scholar] [CrossRef]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of Blood Vessels and Tissues by a Population of Monocytes with Patrolling Behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.M.; Liu, Y.-J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of Monocytes and Dendritic Cells in Blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Wang, X.; Sathe, A.A.; Smith, G.R.; Ruf-Zamojski, F.; Nair, V.; Lavine, K.J.; Xing, C.; Sealfon, S.C.; Zhou, L. Heterogeneous Origins and Functions of Mouse Skeletal Muscle-Resident Macrophages. Proc. Natl. Acad. Sci. USA 2020, 117, 20729–20740. [Google Scholar] [CrossRef]
- Krasniewski, L.K.; Chakraborty, P.; Cui, C.-Y.; Mazan-Mamczarz, K.; Dunn, C.; Piao, Y.; Fan, J.; Shi, C.; Wallace, T.; Nguyen, C.; et al. Single-Cell Analysis of Skeletal Muscle Macrophages Reveals Age-Associated Functional Subpopulations. eLife 2022, 11, e77974. [Google Scholar] [CrossRef]
- Babaeijandaghi, F.; Cheng, R.; Kajabadi, N.; Soliman, H.; Chang, C.-K.; Smandych, J.; Tung, L.W.; Long, R.; Ghassemi, A.; Rossi, F.M.V. Metabolic Reprogramming of Skeletal Muscle by Resident Macrophages Points to CSF1R Inhibitors as Muscular Dystrophy Therapeutics. Sci. Transl. Med. 2022, 14, eabg7504. [Google Scholar] [CrossRef]
- Contreras-Shannon, V.; Ochoa, O.; Reyes-Reyna, S.M.; Sun, D.; Michalek, J.E.; Kuziel, W.A.; McManus, L.M.; Shireman, P.K. Fat Accumulation with Altered Inflammation and Regeneration in Skeletal Muscle of CCR2-/- Mice Following Ischemic Injury. Am. J. Physiol. Cell Physiol. 2007, 292, C953–C967. [Google Scholar] [CrossRef]
- Sun, D.; Martinez, C.O.; Ochoa, O.; Ruiz-Willhite, L.; Bonilla, J.R.; Centonze, V.E.; Waite, L.L.; Michalek, J.E.; McManus, L.M.; Shireman, P.K. Bone Marrow-Derived Cell Regulation of Skeletal Muscle Regeneration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 382–395. [Google Scholar] [CrossRef]
- Lu, H.; Huang, D.; Ransohoff, R.M.; Zhou, L. Acute Skeletal Muscle Injury: CCL2 Expression by Both Monocytes and Injured Muscle Is Required for Repair. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 3344–3355. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Huang, D.; Saederup, N.; Charo, I.F.; Ransohoff, R.M.; Zhou, L. Macrophages Recruited via CCR2 Produce Insulin-like Growth Factor-1 to Repair Acute Skeletal Muscle Injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Melton, D.W.; Porter, L.; Sarwar, Z.U.; McManus, L.M.; Shireman, P.K. Altered Macrophage Phenotype Transition Impairs Skeletal Muscle Regeneration. Am. J. Pathol. 2014, 184, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Dort, J.; Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages Are Key Regulators of Stem Cells during Skeletal Muscle Regeneration and Diseases. Stem Cells Int. 2019, 2019, 4761427. [Google Scholar] [CrossRef] [PubMed]
- McLennan, I.S. Resident Macrophages (ED2- and ED3-Positive) Do Not Phagocytose Degenerating Rat Skeletal Muscle Fibres. Cell Tissue Res. 1993, 272, 193–196. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, L. The Many Roles of Macrophages in Skeletal Muscle Injury and Repair. Front. Cell Dev. Biol. 2022, 10, 952249. [Google Scholar] [CrossRef]
- Brigitte, M.; Schilte, C.; Plonquet, A.; Baba-Amer, Y.; Henri, A.; Charlier, C.; Tajbakhsh, S.; Albert, M.; Gherardi, R.K.; Chrétien, F. Muscle Resident Macrophages Control the Immune Cell Reaction in a Mouse Model of Notexin-Induced Myoinjury. Arthritis Rheum. 2010, 62, 268–279. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in Inflammation, Repair and Regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Varga, T.; Mounier, R.; Gogolak, P.; Poliska, S.; Chazaud, B.; Nagy, L. Tissue LyC6- Macrophages Are Generated in the Absence of Circulating LyC6- Monocytes and Nur77 in a Model of Muscle Regeneration. J. Immunol. 2013, 191, 5695–5701. [Google Scholar] [CrossRef]
- Novak, M.L.; Weinheimer-Haus, E.M.; Koh, T.J. Macrophage Activation and Skeletal Muscle Healing Following Traumatic Injury. J. Pathol. 2014, 232, 344–355. [Google Scholar] [CrossRef]
- Varga, T.; Mounier, R.; Horvath, A.; Cuvellier, S.; Dumont, F.; Poliska, S.; Ardjoune, H.; Juban, G.; Nagy, L.; Chazaud, B. Highly Dynamic Transcriptional Signature of Distinct Macrophage Subsets during Sterile Inflammation, Resolution, and Tissue Repair. J. Immunol. 2016, 196, 4771–4782. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, W.; Ransohoff, R.M.; Zhou, L. Infiltrating Macrophages Are Broadly Activated at the Early Stage to Support Acute Skeletal Muscle Injury Repair. J. Neuroimmunol. 2018, 317, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Villalta, S.A. Regulatory Interactions between Muscle and the Immune System during Muscle Regeneration. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G. Mechanisms of Muscle Injury, Repair, and Regeneration. Compr. Physiol. 2011, 1, 2029–2062. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in Macrophage Phenotypes and Macrophage Competition for Arginine Metabolism Affect the Severity of Muscle Pathology in Muscular Dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef]
- Zhang, J.; Qu, C.; Li, T.; Cui, W.; Wang, X.; Du, J. Phagocytosis Mediated by Scavenger Receptor Class BI Promotes Macrophage Transition during Skeletal Muscle Regeneration. J. Biol. Chem. 2019, 294, 15672–15685. [Google Scholar] [CrossRef]
- Saclier, M.; Lapi, M.; Bonfanti, C.; Rossi, G.; Antonini, S.; Messina, G. The Transcription Factor Nfix Requires RhoA-ROCK1 Dependent Phagocytosis to Mediate Macrophage Skewing during Skeletal Muscle Regeneration. Cells 2020, 9, 708. [Google Scholar] [CrossRef]
- Perdiguero, E.; Sousa-Victor, P.; Ruiz-Bonilla, V.; Jardí, M.; Caelles, C.; Serrano, A.L.; Muñoz-Cánoves, P. P38/MKP-1-Regulated AKT Coordinates Macrophage Transitions and Resolution of Inflammation during Tissue Repair. J. Cell Biol. 2011, 195, 307–322. [Google Scholar] [CrossRef]
- Mounier, R.; Théret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 Regulates Macrophage Skewing at the Time of Resolution of Inflammation during Skeletal Muscle Regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef]
- Baht, G.S.; Bareja, A.; Lee, D.E.; Rao, R.R.; Huang, R.; Huebner, J.L.; Bartlett, D.B.; Hart, C.R.; Gibson, J.R.; Lanza, I.R.; et al. Meteorin-like Facilitates Skeletal Muscle Repair through a Stat3/IGF-1 Mechanism. Nat. Metab. 2020, 2, 278–289. [Google Scholar] [CrossRef]
- Tonkin, J.; Temmerman, L.; Sampson, R.D.; Gallego-Colon, E.; Barberi, L.; Bilbao, D.; Schneider, M.D.; Musarò, A.; Rosenthal, N. Monocyte/Macrophage-Derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, D.; Mourkioti, F.; Gambardella, A.; Kirstetter, P.; Lopez, R.G.; Rosenthal, N.; Nerlov, C. A CREB-C/EBPbeta Cascade Induces M2 Macrophage-Specific Gene Expression and Promotes Muscle Injury Repair. Proc. Natl. Acad. Sci. USA 2009, 106, 17475–17480. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Savill, J. Resolution of Inflammation: The Beginning Programs the End. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Borrego, I.; Frobert, A.; Ajalbert, G.; Valentin, J.; Kaltenrieder, C.; Fellay, B.; Stumpe, M.; Cook, S.; Dengjel, J.; Giraud, M.-N. Fibrin, Bone Marrow Cells and Macrophages Interactively Modulate Cardiomyoblast Fate. Biomedicines 2022, 10, 527. [Google Scholar] [CrossRef]
- Hernandez-Torres, F.; Rodríguez-Outeiriño, L.; Franco, D.; Aranega, A.E. Pitx2 in Embryonic and Adult Myogenesis. Front. Cell Dev. Biol. 2017, 5, 46. [Google Scholar] [CrossRef]
- Lepper, C.; Partridge, T.A.; Fan, C.-M. An Absolute Requirement for Pax7-Positive Satellite Cells in Acute Injury-Induced Skeletal Muscle Regeneration. Dev. Camb. Engl. 2011, 138, 3639–3646. [Google Scholar] [CrossRef]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite Cells, Connective Tissue Fibroblasts and Their Interactions Are Crucial for Muscle Regeneration. Dev. Camb. Engl. 2011, 138, 3625–3637. [Google Scholar] [CrossRef]
- Sambasivan, R.; Yao, R.; Kissenpfennig, A.; Van Wittenberghe, L.; Paldi, A.; Gayraud-Morel, B.; Guenou, H.; Malissen, B.; Tajbakhsh, S.; Galy, A. Pax7-Expressing Satellite Cells Are Indispensable for Adult Skeletal Muscle Regeneration. Dev. Camb. Engl. 2011, 138, 3647–3656. [Google Scholar] [CrossRef]
- Mauro, A. Satellite Cell of Skeletal Muscle Fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 Is Required for the Specification of Myogenic Satellite Cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Crist, C.G.; Montarras, D.; Buckingham, M. Muscle Satellite Cells Are Primed for Myogenesis but Maintain Quiescence with Sequestration of Myf5 mRNA Targeted by microRNA-31 in mRNP Granules. Cell Stem Cell 2012, 11, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Kuroda, K.; Le Grand, F.; Rudnicki, M.A. Asymmetric Self-Renewal and Commitment of Satellite Stem Cells in Muscle. Cell 2007, 129, 999–1010. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Dumont, N.A.; Rudnicki, M.A. Cellular Dynamics in the Muscle Satellite Cell Niche. EMBO Rep. 2013, 14, 1062–1072. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite Cells and the Muscle Stem Cell Niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.-C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef]
- Chazaud, B.; Sonnet, C.; Lafuste, P.; Bassez, G.; Rimaniol, A.-C.; Poron, F.; Authier, F.-J.; Dreyfus, P.A.; Gherardi, R.K. Satellite Cells Attract Monocytes and Use Macrophages as a Support to Escape Apoptosis and Enhance Muscle Growth. J. Cell Biol. 2003, 163, 1133–1143. [Google Scholar] [CrossRef]
- Sonnet, C.; Lafuste, P.; Arnold, L.; Brigitte, M.; Poron, F.; Authier, F.-J.; Chrétien, F.; Gherardi, R.K.; Chazaud, B. Human Macrophages Rescue Myoblasts and Myotubes from Apoptosis through a Set of Adhesion Molecular Systems. J. Cell Sci. 2006, 119, 2497–2507. [Google Scholar] [CrossRef]
- Juhas, M.; Abutaleb, N.; Wang, J.T.; Ye, J.; Shaikh, Z.; Sriworarat, C.; Qian, Y.; Bursac, N. Incorporation of Macrophages into Engineered Skeletal Muscle Enables Enhanced Muscle Regeneration. Nat. Biomed. Eng. 2018, 2, 942–954. [Google Scholar] [CrossRef]
- Li, Y.-P. TNF-Alpha Is a Mitogen in Skeletal Muscle. Am. J. Physiol. Cell Physiol. 2003, 285, C370–C376. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Y.; Wu, Y.; Wang, L.; Wang, X.; Du, J. Interleukin-6/Signal Transducer and Activator of Transcription 3 (STAT3) Pathway Is Essential for Macrophage Infiltration and Myoblast Proliferation during Muscle Regeneration. J. Biol. Chem. 2013, 288, 1489–1499. [Google Scholar] [CrossRef]
- Ho, A.T.V.; Palla, A.R.; Blake, M.R.; Yucel, N.D.; Wang, Y.X.; Magnusson, K.E.G.; Holbrook, C.A.; Kraft, P.E.; Delp, S.L.; Blau, H.M. Prostaglandin E2 Is Essential for Efficacious Skeletal Muscle Stem-Cell Function, Augmenting Regeneration and Strength. Proc. Natl. Acad. Sci. USA 2017, 114, 6675–6684. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Shih, C.-H.; Wosczyna, M.N.; Mueller, A.A.; Cho, J.; Aggarwal, A.; Rando, T.A.; Feldman, B.J. Macrophage-Released ADAMTS1 Promotes Muscle Stem Cell Activation. Nat. Commun. 2017, 8, 669. [Google Scholar] [CrossRef] [PubMed]
- Al-Shanti, N.; Saini, A.; Faulkner, S.H.; Stewart, C.E. Beneficial Synergistic Interactions of TNF-Alpha and IL-6 in C2 Skeletal Myoblasts—Potential Cross-Talk with IGF System. Growth Factors 2008, 26, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Yuasa, S.; Shimoji, K.; Onizuka, T.; Hayashiji, N.; Ohno, Y.; Arai, T.; Hattori, F.; Kaneda, R.; Kimura, K.; et al. G-CSF Influences Mouse Skeletal Muscle Development and Regeneration by Stimulating Myoblast Proliferation. J. Exp. Med. 2011, 208, 715–727. [Google Scholar] [CrossRef]
- Horsley, V.; Jansen, K.M.; Mills, S.T.; Pavlath, G.K. IL-4 Acts as a Myoblast Recruitment Factor during Mammalian Muscle Growth. Cell 2003, 113, 483–494. [Google Scholar] [CrossRef]
- Dumont, N.; Frenette, J. Macrophages Protect against Muscle Atrophy and Promote Muscle Recovery in Vivo and in Vitro: A Mechanism Partly Dependent on the Insulin-like Growth Factor-1 Signaling Molecule. Am. J. Pathol. 2010, 176, 2228–2235. [Google Scholar] [CrossRef]
- Latroche, C.; Weiss-Gayet, M.; Muller, L.; Gitiaux, C.; Leblanc, P.; Liot, S.; Ben-Larbi, S.; Abou-Khalil, R.; Verger, N.; Bardot, P.; et al. Coupling between Myogenesis and Angiogenesis during Skeletal Muscle Regeneration Is Stimulated by Restorative Macrophages. Stem Cell Rep. 2017, 9, 2018–2033. [Google Scholar] [CrossRef]
- Varga, T.; Mounier, R.; Patsalos, A.; Gogolák, P.; Peloquin, M.; Horvath, A.; Pap, A.; Daniel, B.; Nagy, G.; Pintye, E.; et al. Macrophage PPARγ, a Lipid Activated Transcription Factor Controls the Growth Factor GDF3 and Skeletal Muscle Regeneration. Immunity 2016, 45, 1038–1051. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Zhang, H. Extracellular Matrix: An Important Regulator of Cell Functions and Skeletal Muscle Development. Cell Biosci. 2021, 11, 65. [Google Scholar] [CrossRef]
- Huang, G.; Ge, G.; Wang, D.; Gopalakrishnan, B.; Butz, D.H.; Colman, R.J.; Nagy, A.; Greenspan, D.S. A3(V) Collagen Is Critical for Glucose Homeostasis in Mice Due to Effects in Pancreatic Islets and Peripheral Tissues. J. Clin. Investig. 2011, 121, 769–783. [Google Scholar] [CrossRef]
- Joe, A.W.B.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M.V. Muscle Injury Activates Resident Fibro/Adipogenic Progenitors That Facilitate Myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 Innate Signals Stimulate Fibro/Adipogenic Progenitors to Facilitate Muscle Regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Loomis, T.; Hu, L.-Y.; Wohlgemuth, R.P.; Chellakudam, R.R.; Muralidharan, P.D.; Smith, L.R. Matrix Stiffness and Architecture Drive Fibro-Adipogenic Progenitors’ Activation into Myofibroblasts. Sci. Rep. 2022, 12, 13582. [Google Scholar] [CrossRef] [PubMed]
- Uezumi, A.; Ikemoto-Uezumi, M.; Tsuchida, K. Roles of Nonmyogenic Mesenchymal Progenitors in Pathogenesis and Regeneration of Skeletal Muscle. Front. Physiol. 2014, 5, 68. [Google Scholar] [CrossRef]
- Lemos, D.R.; Babaeijandaghi, F.; Low, M.; Chang, C.-K.; Lee, S.T.; Fiore, D.; Zhang, R.-H.; Natarajan, A.; Nedospasov, S.A.; Rossi, F.M.V. Nilotinib Reduces Muscle Fibrosis in Chronic Muscle Injury by Promoting TNF-Mediated Apoptosis of Fibro/Adipogenic Progenitors. Nat. Med. 2015, 21, 786–794. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, W.; Ransohoff, R.M.; Zhou, L. Identification and Function of Fibrocytes in Skeletal Muscle Injury Repair and Muscular Dystrophy. J. Immunol. 2016, 197, 4750–4761. [Google Scholar] [CrossRef]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-Based Path to Newborn Screening for Duchenne Muscular Dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef]
- Tavakoli, N.P.; Gruber, D.; Armstrong, N.; Chung, W.K.; Maloney, B.; Park, S.; Wynn, J.; Koval-Burt, C.; Verdade, L.; Tegay, D.H.; et al. Newborn Screening for Duchenne Muscular Dystrophy: A Two-year Pilot Study. Ann. Clin. Transl. Neurol. 2023, 10, 1383–1396. [Google Scholar] [CrossRef]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef]
- Stark, A.E. Determinants of the Incidence of Duchenne Muscular Dystrophy. Ann. Transl. Med. 2015, 3, 287. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F. Muscular Dystrophies. Lancet 2013, 381, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Gervasio, O.L.; Yeung, E.W.; Whitehead, N.P. Calcium and the Damage Pathways in Muscular Dystrophy. Can. J. Physiol. Pharmacol. 2010, 88, 83–91. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin Expression in Muscle Stem Cells Regulates Their Polarity and Asymmetric Division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short Telomeres and Stem Cell Exhaustion Model Duchenne Muscular Dystrophy in Mdx/mTR Mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef]
- Webster, C.; Blau, H.M. Accelerated Age-Related Decline in Replicative Life-Span of Duchenne Muscular Dystrophy Myoblasts: Implications for Cell and Gene Therapy. Somat. Cell Mol. Genet. 1990, 16, 557–565. [Google Scholar] [CrossRef]
- Tidball, J.G.; Wehling-Henricks, M. Evolving Therapeutic Strategies for Duchenne Muscular Dystrophy: Targeting Downstream Events. Pediatr. Res. 2004, 56, 831–841. [Google Scholar] [CrossRef]
- Manzur, A.Y.; Kuntzer, T.; Pike, M.; Swan, A. Glucocorticoid Corticosteroids for Duchenne Muscular Dystrophy. Cochrane Database Syst. Rev. 2008, CD003725. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Neuromuscular, Rehabilitation, Endocrine, and Gastrointestinal and Nutritional Management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-Term Effects of Glucocorticoids on Function, Quality of Life, and Survival in Patients with Duchenne Muscular Dystrophy: A Prospective Cohort Study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.-W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous Remodeling Is a Driver of Failed Regeneration in Duchenne Muscular Dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef]
- Villalta, S.A.; Rinaldi, C.; Deng, B.; Liu, G.; Fedor, B.; Tidball, J.G. Interleukin-10 Reduces the Pathology of Mdx Muscular Dystrophy by Deactivating M1 Macrophages and Modulating Macrophage Phenotype. Hum. Mol. Genet. 2011, 20, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Jordan, M.C.; Gotoh, T.; Grody, W.W.; Roos, K.P.; Tidball, J.G. Arginine Metabolism by Macrophages Promotes Cardiac and Muscle Fibrosis in Mdx Muscular Dystrophy. PLoS ONE 2010, 5, e10763. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic Switching of Adipose Tissue Macrophages with Obesity Is Generated by Spatiotemporal Differences in Macrophage Subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage Activation and Polarization. Front. Biosci. J. Virtual Libr. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 Activation of Kupffer Cells by PPARdelta Ameliorates Obesity-Induced Insulin Resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef]
- Rybarczyk, B.J.; Lawrence, S.O.; Simpson-Haidaris, P.J. Matrix-Fibrinogen Enhances Wound Closure by Increasing Both Cell Proliferation and Migration. Blood 2003, 102, 4035–4043. [Google Scholar] [CrossRef]
- Vidal, B.; Serrano, A.L.; Tjwa, M.; Suelves, M.; Ardite, E.; De Mori, R.; Baeza-Raja, B.; Martínez de Lagrán, M.; Lafuste, P.; Ruiz-Bonilla, V.; et al. Fibrinogen Drives Dystrophic Muscle Fibrosis via a TGFbeta/Alternative Macrophage Activation Pathway. Genes Dev. 2008, 22, 1747–1752. [Google Scholar] [CrossRef]
- Juban, G.; Saclier, M.; Yacoub-Youssef, H.; Kernou, A.; Arnold, L.; Boisson, C.; Ben Larbi, S.; Magnan, M.; Cuvellier, S.; Théret, M.; et al. AMPK Activation Regulates LTBP4-Dependent TGF-Β1 Secretion by Pro-Inflammatory Macrophages and Controls Fibrosis in Duchenne Muscular Dystrophy. Cell Rep. 2018, 25, 2163–2176.e6. [Google Scholar] [CrossRef]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.L.; Carathers, M.; Li, Z.-W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NF-kappaB Signaling in Macrophages and Myofibers Promotes Muscle Degeneration in Duchenne Muscular Dystrophy. J. Clin. Investig. 2007, 117, 889–901. [Google Scholar] [CrossRef]
- Mojumdar, K.; Liang, F.; Giordano, C.; Lemaire, C.; Danialou, G.; Okazaki, T.; Bourdon, J.; Rafei, M.; Galipeau, J.; Divangahi, M.; et al. Inflammatory Monocytes Promote Progression of Duchenne Muscular Dystrophy and Can Be Therapeutically Targeted via CCR2. EMBO Mol. Med. 2014, 6, 1476–1492. [Google Scholar] [CrossRef]
- Rizzo, G.; Di Maggio, R.; Benedetti, A.; Morroni, J.; Bouche, M.; Lozanoska-Ochser, B. Splenic Ly6Chi Monocytes Are Critical Players in Dystrophic Muscle Injury and Repair. JCI Insight 2020, 5, e130807. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Mojumdar, K.; Liang, F.; Lemaire, C.; Li, T.; Richardson, J.; Divangahi, M.; Qureshi, S.; Petrof, B.J. Toll-like Receptor 4 Ablation in Mdx Mice Reveals Innate Immunity as a Therapeutic Target in Duchenne Muscular Dystrophy. Hum. Mol. Genet. 2015, 24, 2147–2162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Porter, J.D.; Cheng, G.; Gong, B.; Hatala, D.A.; Merriam, A.P.; Zhou, X.; Rafael, J.A.; Kaminski, H.J. Temporal and Spatial mRNA Expression Patterns of TGF-Beta1, 2, 3 and TbetaRI, II, III in Skeletal Muscles of Mdx Mice. Neuromuscul. Disord. NMD 2006, 16, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Saleh, K.K.; Xi, H.; Switzler, C.; Skuratovsky, E.; Romero, M.A.; Chien, P.; Gibbs, D.; Gane, L.; Hicks, M.R.; Spencer, M.J.; et al. Single Cell Sequencing Maps Skeletal Muscle Cellular Diversity as Disease Severity Increases in Dystrophic Mouse Models. iScience 2022, 25, 105415. [Google Scholar] [CrossRef]
- Bhattarai, S.; Li, Q.; Ding, J.; Liang, F.; Gusev, E.; Lapohos, O.; Fonseca, G.J.; Kaufmann, E.; Divangahi, M.; Petrof, B.J. TLR4 Is a Regulator of Trained Immunity in a Murine Model of Duchenne Muscular Dystrophy. Nat. Commun. 2022, 13, 879. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; van der Meer, J.W.M. Trained Immunity: A Memory for Innate Host Defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-Specific Control of Inflammation by TLR-Induced Chromatin Modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef]
- Netea, M.G.; Schlitzer, A.; Placek, K.; Joosten, L.A.B.; Schultze, J.L. Innate and Adaptive Immune Memory: An Evolutionary Continuum in the Host’s Response to Pathogens. Cell Host Microbe 2019, 25, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Divangahi, M.; Aaby, P.; Khader, S.A.; Barreiro, L.B.; Bekkering, S.; Chavakis, T.; van Crevel, R.; Curtis, N.; DiNardo, A.R.; Dominguez-Andres, J.; et al. Trained Immunity, Tolerance, Priming and Differentiation: Distinct Immunological Processes. Nat. Immunol. 2021, 22, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Davies, K.E. Pharmacological Advances for Treatment in Duchenne Muscular Dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Moxley, R.T.; Pandya, S.; Ciafaloni, E.; Fox, D.J.; Campbell, K. Change in Natural History of Duchenne Muscular Dystrophy with Long-Term Corticosteroid Treatment: Implications for Management. J. Child Neurol. 2010, 25, 1116–1129. [Google Scholar] [CrossRef]
- Hussein, M.R.; Hamed, S.A.; Mostafa, M.G.; Abu-Dief, E.E.; Kamel, N.F.; Kandil, M.R. The Effects of Glucocorticoid Therapy on the Inflammatory and Dendritic Cells in Muscular Dystrophies. Int. J. Exp. Pathol. 2006, 87, 451–461. [Google Scholar] [CrossRef]
- Griggs, R.C.; Moxley, R.T.; Mendell, J.R.; Fenichel, G.M.; Brooke, M.H.; Pestronk, A.; Miller, J.P. Prednisone in Duchenne Dystrophy. A Randomized, Controlled Trial Defining the Time Course and Dose Response. Clinical Investigation of Duchenne Dystrophy Group. Arch. Neurol. 1991, 48, 383–388. [Google Scholar] [CrossRef]
- Joseph, S.; Wang, C.; Bushby, K.; Guglieri, M.; Horrocks, I.; Straub, V.; Ahmed, S.F.; Wong, S.C.; UK NorthStar Clinical Network. Fractures and Linear Growth in a Nationwide Cohort of Boys with Duchenne Muscular Dystrophy with and without Glucocorticoid Treatment: Results From the UK NorthStar Database. JAMA Neurol. 2019, 76, 701–709. [Google Scholar] [CrossRef]
- Bylo, M.; Farewell, R.; Coppenrath, V.A.; Yogaratnam, D. A Review of Deflazacort for Patients With Duchenne Muscular Dystrophy. Ann. Pharmacother. 2020, 54, 788–794. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Zelikovich, A.S.; Salamone, I.M.; Fischer, J.A.; McNally, E.M. Mechanisms and Clinical Applications of Glucocorticoid Steroids in Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, 39–52. [Google Scholar] [CrossRef]
- Wang, F.-S.; Ko, J.-Y.; Weng, L.-H.; Yeh, D.-W.; Ke, H.-J.; Wu, S.-L. Inhibition of Glycogen Synthase Kinase-3beta Attenuates Glucocorticoid-Induced Bone Loss. Life Sci. 2009, 85, 685–692. [Google Scholar] [CrossRef]
- van der Velden, J.L.J.; Langen, R.C.J.; Kelders, M.C.J.M.; Wouters, E.F.M.; Janssen-Heininger, Y.M.W.; Schols, A.M.W.J. Inhibition of Glycogen Synthase Kinase-3beta Activity Is Sufficient to Stimulate Myogenic Differentiation. Am. J. Physiol. Cell Physiol. 2006, 290, C453–C462. [Google Scholar] [CrossRef] [PubMed]
- Schakman, O.; Gilson, H.; Thissen, J.P. Mechanisms of Glucocorticoid-Induced Myopathy. J. Endocrinol. 2008, 197, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Nader, G.A. Balancing Muscle Hypertrophy and Atrophy. Nat. Med. 2004, 10, 584–585. [Google Scholar] [CrossRef]
- Murray, P.J. Understanding and Exploiting the Endogenous Interleukin-10/STAT3-Mediated Anti-Inflammatory Response. Curr. Opin. Pharmacol. 2006, 6, 379–386. [Google Scholar] [CrossRef]
- Mosser, D.M.; Zhang, X. Interleukin-10: New Perspectives on an Old Cytokine. Immunol. Rev. 2008, 226, 205–218. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Lang, R.; Rutschman, R.L.; Greaves, D.R.; Murray, P.J. Autocrine Deactivation of Macrophages in Transgenic Mice Constitutively Overexpressing IL-10 under Control of the Human CD68 Promoter. J. Immunol. 2002, 168, 3402–3411. [Google Scholar] [CrossRef]
- Villalta, S.A.; Deng, B.; Rinaldi, C.; Wehling-Henricks, M.; Tidball, J.G. IFNγ Promotes Muscle Damage in the Mdx Mouse Model of Duchenne Muscular Dystrophy by Suppressing M2 Macrophage Activation and Inhibiting Muscle Cell Proliferation. J. Immunol. 2011, 187, 5419–5428. [Google Scholar] [CrossRef]
- Grounds, M.D.; Radley, H.G.; Gebski, B.L.; Bogoyevitch, M.A.; Shavlakadze, T. Implications of Cross-Talk between Tumour Necrosis Factor and Insulin-like Growth Factor-1 Signalling in Skeletal Muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 846–851. [Google Scholar] [CrossRef]
- Messina, S.; Bitto, A.; Aguennouz, M.; Minutoli, L.; Monici, M.C.; Altavilla, D.; Squadrito, F.; Vita, G. Nuclear Factor Kappa-B Blockade Reduces Skeletal Muscle Degeneration and Enhances Muscle Function in Mdx Mice. Exp. Neurol. 2006, 198, 234–241. [Google Scholar] [CrossRef]
- Kuru, S.; Inukai, A.; Kato, T.; Liang, Y.; Kimura, S.; Sobue, G. Expression of Tumor Necrosis Factor-Alpha in Regenerating Muscle Fibers in Inflammatory and Non-Inflammatory Myopathies. Acta Neuropathol. 2003, 105, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Radley, H.G.; Davies, M.J.; Grounds, M.D. Reduced Muscle Necrosis and Long-Term Benefits in Dystrophic Mdx Mice after cV1q (Blockade of TNF) Treatment. Neuromuscul. Disord. NMD 2008, 18, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Torrisi, J. Anti-TNFalpha (Remicade) Therapy Protects Dystrophic Skeletal Muscle from Necrosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 676–682. [Google Scholar] [CrossRef]
- Hodgetts, S.; Radley, H.; Davies, M.; Grounds, M.D. Reduced Necrosis of Dystrophic Muscle by Depletion of Host Neutrophils, or Blocking TNFalpha Function with Etanercept in Mdx Mice. Neuromuscul. Disord. NMD 2006, 16, 591–602. [Google Scholar] [CrossRef]
- Chan, Y.-S.; Li, Y.; Foster, W.; Horaguchi, T.; Somogyi, G.; Fu, F.H.; Huard, J. Antifibrotic Effects of Suramin in Injured Skeletal Muscle after Laceration. J. Appl. Physiol. 2003, 95, 771–780. [Google Scholar] [CrossRef]
- Chan, Y.-S.; Li, Y.; Foster, W.; Fu, F.H.; Huard, J. The Use of Suramin, an Antifibrotic Agent, to Improve Muscle Recovery after Strain Injury. Am. J. Sports Med. 2005, 33, 43–51. [Google Scholar] [CrossRef]
- Nozaki, M.; Li, Y.; Zhu, J.; Ambrosio, F.; Uehara, K.; Fu, F.H.; Huard, J. Improved Muscle Healing after Contusion Injury by the Inhibitory Effect of Suramin on Myostatin, a Negative Regulator of Muscle Growth. Am. J. Sports Med. 2008, 36, 2354–2362. [Google Scholar] [CrossRef]
- Coffey, R.J.; Leof, E.B.; Shipley, G.D.; Moses, H.L. Suramin Inhibition of Growth Factor Receptor Binding and Mitogenicity in AKR-2B Cells. J. Cell. Physiol. 1987, 132, 143–148. [Google Scholar] [CrossRef]
- La Rocca, R.V.; Stein, C.A.; Danesi, R.; Myers, C.E. Suramin, a Novel Antitumor Compound. J. Steroid Biochem. Mol. Biol. 1990, 37, 893–898. [Google Scholar] [CrossRef]
- McGeary, R.P.; Bennett, A.J.; Tran, Q.B.; Cosgrove, K.L.; Ross, B.P. Suramin: Clinical Uses and Structure-Activity Relationships. Mini Rev. Med. Chem. 2008, 8, 1384–1394. [Google Scholar] [CrossRef]
- Taniguti, A.P.T.; Pertille, A.; Matsumura, C.Y.; Santo Neto, H.; Marques, M.J. Prevention of Muscle Fibrosis and Myonecrosis in Mdx Mice by Suramin, a TGF-Β1 Blocker. Muscle Nerve 2011, 43, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From Mechanisms of Action to Therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Guigas, B.; Viollet, B. Targeting AMPK: From Ancient Drugs to New Small-Molecule Activators. In AMP-Activated Protein Kinase; Experientia Supplementum; Springer: Cham, Switzerland, 2016; Volume 107, pp. 327–350. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-Stimulating Factor 1 Receptor (CSF1R) Inhibitors in Cancer Therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Cassier, P.A.; Italiano, A.; Gomez-Roca, C.; Le Tourneau, C.; Toulmonde, M.; D’Angelo, S.P.; Weber, K.; Loirat, D.; Jacob, W.; Jegg, A.-M.; et al. Long-Term Clinical Activity, Safety and Patient-Reported Quality of Life for Emactuzumab-Treated Patients with Diffuse-Type Tenosynovial Giant-Cell Tumour. Eur. J. Cancer 2020, 141, 162–170. [Google Scholar] [CrossRef]
- Sauter, K.A.; Pridans, C.; Sehgal, A.; Tsai, Y.T.; Bradford, B.M.; Raza, S.; Moffat, L.; Gow, D.J.; Beard, P.M.; Mabbott, N.A.; et al. Pleiotropic Effects of Extended Blockade of CSF1R Signaling in Adult Mice. J. Leukoc. Biol. 2014, 96, 265–274. [Google Scholar] [CrossRef]
- Saclier, M.; Angelini, G.; Bonfanti, C.; Mura, G.; Temponi, G.; Messina, G. Selective Ablation of Nfix in Macrophages Attenuates Muscular Dystrophy by Inhibiting Fibro-Adipogenic Progenitor-Dependent Fibrosis. J. Pathol. 2022, 257, 352–366. [Google Scholar] [CrossRef]
- Gronostajski, R.M. Roles of the NFI/CTF Gene Family in Transcription and Development. Gene 2000, 249, 31–45. [Google Scholar] [CrossRef]
- Rossi, G.; Bonfanti, C.; Antonini, S.; Bastoni, M.; Monteverde, S.; Innocenzi, A.; Saclier, M.; Taglietti, V.; Messina, G. Silencing Nfix Rescues Muscular Dystrophy by Delaying Muscle Regeneration. Nat. Commun. 2017, 8, 1055. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF-κB Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Miyatake, S.; Shimizu-Motohashi, Y.; Takeda, S.; Aoki, Y. Anti-Inflammatory Drugs for Duchenne Muscular Dystrophy: Focus on Skeletal Muscle-Releasing Factors. Drug Des. Devel. Ther. 2016, 10, 2745–2758. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, K.A.; Wilson, W.; Cogswell, P.C.; Baldwin, A.S. Glycogen Synthase Kinase 3β Functions To Specify Gene-Specific, NF-κB-Dependent Transcription. Mol. Cell. Biol. 2005, 25, 8444–8455. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for Glycogen Synthase Kinase-3beta in Cell Survival and NF-kappaB Activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like Receptor—Mediated Cytokine Production Is Differentially Regulated by Glycogen Synthase Kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Wang, H.; Brown, J.; Gu, Z.; Garcia, C.A.; Liang, R.; Alard, P.; Beurel, E.; Jope, R.S.; Greenway, T.; Martin, M. Convergence of the Mammalian Target of Rapamycin Complex 1- and Glycogen Synthase Kinase 3-β–Signaling Pathways Regulates the Innate Inflammatory Response. J. Immunol. 2011, 186, 5217–5226. [Google Scholar] [CrossRef]
- Matias-Valiente, L.; Sanchez-Fernandez, C.; Rodriguez-Outeiriño, L.; Ramos, M.C.; Díaz, C.; Crespo, G.; González-Menéndez, V.; Genilloud, O.; Reyes, F.; Montolio, M.; et al. Evaluation of Pro-Regenerative and Anti-Inflammatory Effects of Isolecanoric Acid in the Muscle: Potential Treatment of Duchenne Muscular Dystrophy. Biomed. Pharmacother. 2023, 170, 116056. [Google Scholar] [CrossRef]
- de Pedro, N.; Cantizani, J.; Ortiz-López, F.J.; González-Menéndez, V.; Cautain, B.; Rodríguez, L.; Bills, G.F.; Reyes, F.; Genilloud, O.; Vicente, F. Protective Effects of Isolecanoric Acid on Neurodegenerative in Vitro Models. Neuropharmacology 2016, 101, 538–548. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a Late Mediator of Endotoxin Lethality in Mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The Nuclear Protein HMGB1 Is Secreted by Monocytes via a Non-Classical, Vesicle-Mediated Secretory Pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez-Torres, F.; Matias-Valiente, L.; Alzas-Gomez, V.; Aranega, A.E. Macrophages in the Context of Muscle Regeneration and Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2024, 25, 10393. https://doi.org/10.3390/ijms251910393
Hernandez-Torres F, Matias-Valiente L, Alzas-Gomez V, Aranega AE. Macrophages in the Context of Muscle Regeneration and Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2024; 25(19):10393. https://doi.org/10.3390/ijms251910393
Chicago/Turabian StyleHernandez-Torres, Francisco, Lidia Matias-Valiente, Virginia Alzas-Gomez, and Amelia Eva Aranega. 2024. "Macrophages in the Context of Muscle Regeneration and Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 25, no. 19: 10393. https://doi.org/10.3390/ijms251910393
APA StyleHernandez-Torres, F., Matias-Valiente, L., Alzas-Gomez, V., & Aranega, A. E. (2024). Macrophages in the Context of Muscle Regeneration and Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 25(19), 10393. https://doi.org/10.3390/ijms251910393