

Biased Signaling Agonists Promote Distinct Phosphorylation and Conformational States of the Dopamine D3 Receptor


Abstract
:1. Introduction
2. Results
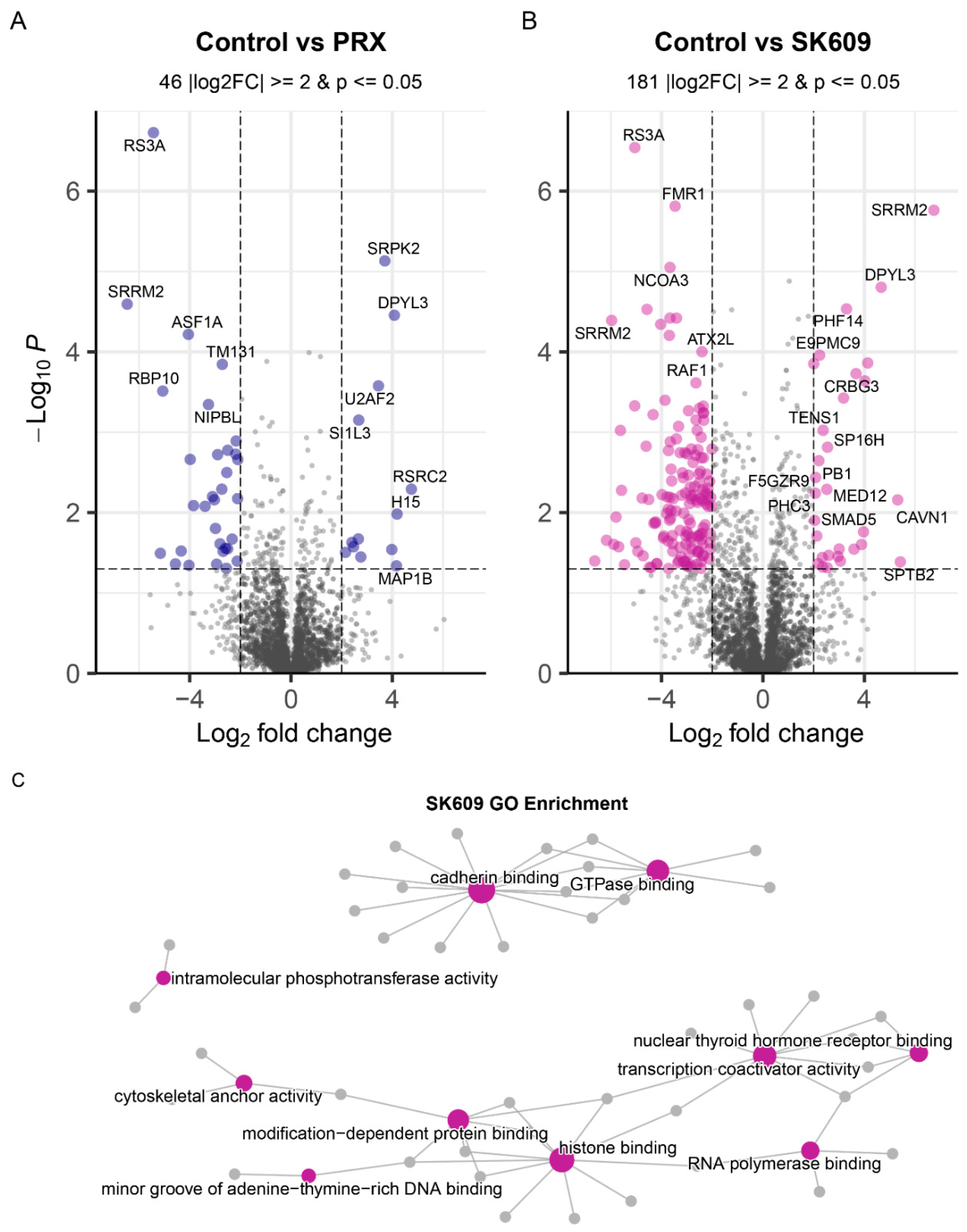
2.1. PRX and SK609 Treatment May Promote Different Phosphorylation States of D3R
2.2. PRX and SK609 Impart Differential Pathway Utilization
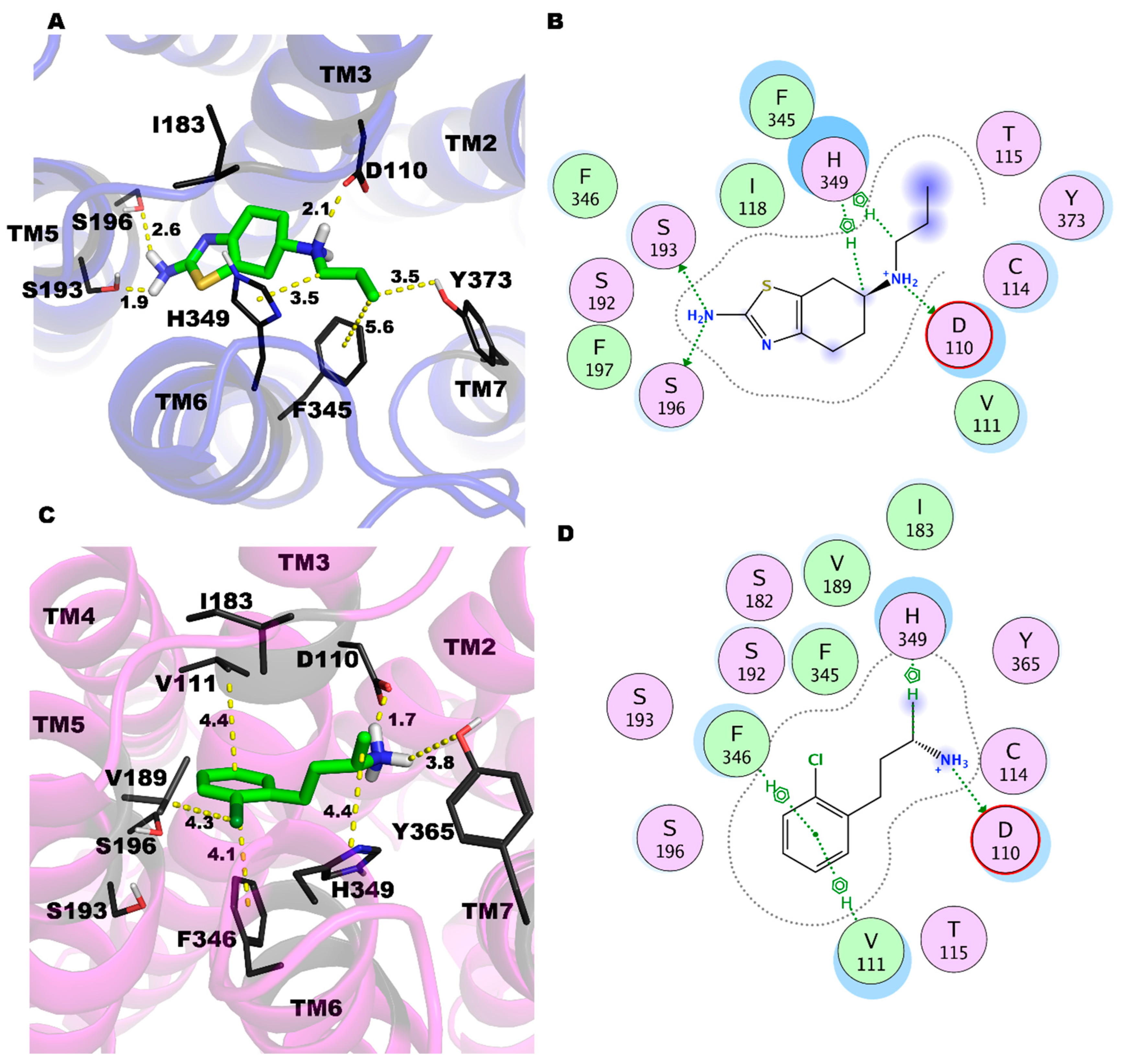
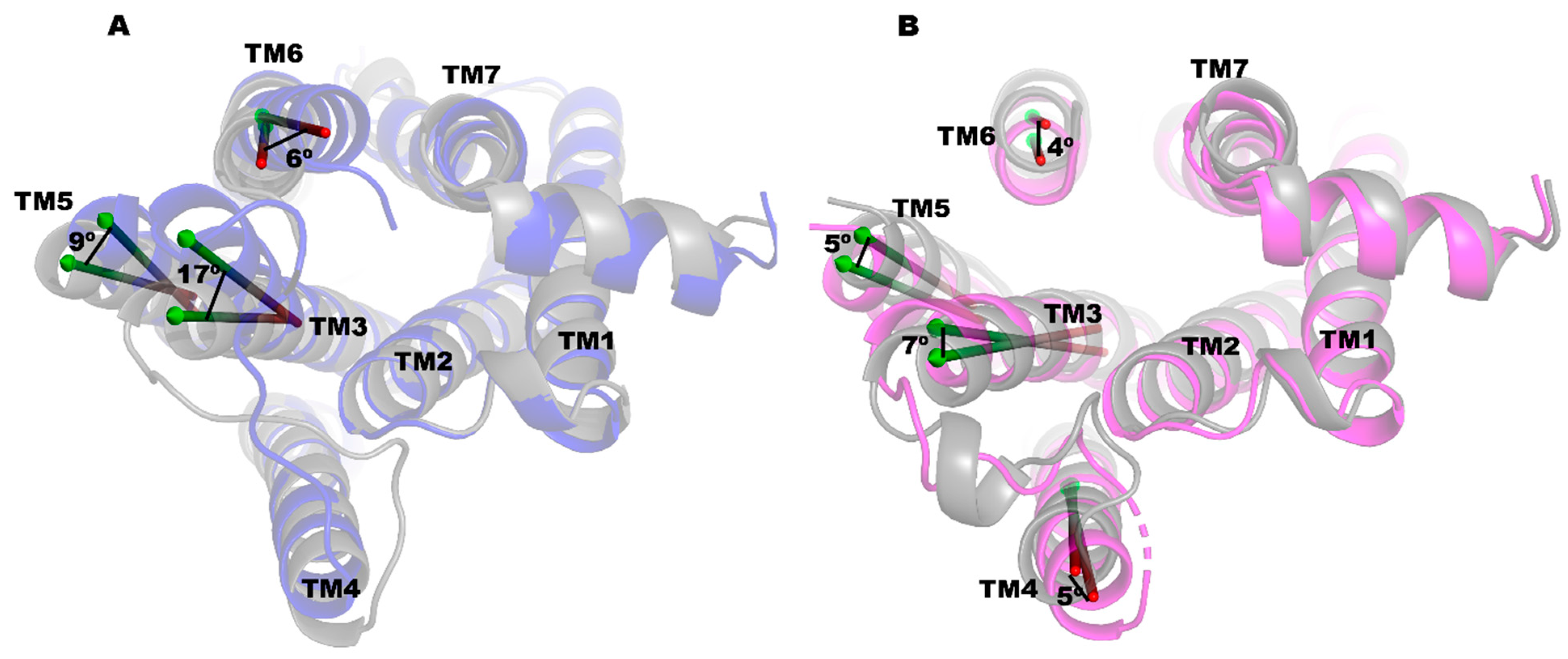
2.3. PRX and SK609 Promote Different Conformational Changes in D3R
2.4. D3R-βarr2-a Complexes of PRX and SK609 Do Not Support Experimental Findings
2.5. D3R-βarr2-b Complexes Are More Plausible than the D3R-βarr2-a Complexes
2.6. The Conformation of the Double-Mutant D3R (Leu121Asn and Tyr129Leu)-PRX Complex Is More Homologous to the D3R-SK609 Complex
2.7. PRX- and SK609-Bound D3R-Gi Complexes Have Similar Conformations
3. Discussion
4. Methods
4.1. Untargeted Phosphoproteomics
4.2. Pathway and GO Enrichment Analysis
4.3. Molecular Modeling and MD Simulations
4.4. Modeling of the PRX- and SK609-Bound D3R-βarr2 Complexes
4.5. Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heng, B.C.; Aubel, D.; Fussenegger, M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 2013, 31, 1676–1694. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G protein-coupled receptors: Structure- and function-based drug discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J. G-protein-coupled receptors: Past, present and future. Br. J. Pharmacol. 2006, 147, S27–S37. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. An Overview on GPCRs and Drug Discovery: Structure-Based Drug Design and Structural Biology on GPCRs. In G Protein-Coupled Receptors in Drug Discovery; Leifert, W.R., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 51–66. [Google Scholar]
- Garland, S.L. Are GPCRs Still a Source of New Targets? J. Biomol. Screen. 2013, 18, 947–966. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Xiao, K.; Lefkowitz, R.J. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 2011, 36, 457–469. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased Opioid Ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G Protein-Biased Ligand at the μ-Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef]
- Slosky, L.M.; Bai, Y.; Toth, K.; Ray, C.; Rochelle, L.K.; Badea, A.; Chandrasekhar, R.; Pogorelov, V.M.; Abraham, D.M.; Atluri, N.; et al. β-Arrestin-Biased Allosteric Modulator of NTSR1 Selectively Attenuates Addictive Behaviors. Cell 2020, 181, 1364–1379.e14. [Google Scholar] [CrossRef]
- Urs, N.M.; Bido, S.; Peterson, S.M.; Daigle, T.L.; Bass, C.E.; Gainetdinov, R.R.; Bezard, E.; Caron, M.G. Targeting β-arrestin2 in the treatment of l-DOPA–induced dyskinesia in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2015, 112, E2517–E2526. [Google Scholar] [CrossRef]
- Inui, S. Nalfurafine hydrochloride to treat pruritus: A review. Clin. Cosmet. Investig. Dermatol. 2015, 8, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-Based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Wisler, J.W.; DeWire, S.M.; Whalen, E.J.; Violin, J.D.; Drake, M.T.; Ahn, S.; Shenoy, S.K.; Lefkowitz, R.J. A unique mechanism of β-blocker action: Carvedilol stimulates β-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar] [CrossRef]
- Miyano, K.; Manabe, S.; Komatsu, A.; Fujii, Y.; Mizobuchi, Y.; Uezono, E.; Ohshima, K.; Nonaka, M.; Kuroda, Y.; Narita, M.; et al. The G Protein Signal-Biased Compound TRV130; Structures, Its Site of Action and Clinical Studies. Curr. Top. Med. Chem. 2020, 20, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Viswanathan, G.; Sellig, M.; Jassal, C.; Choi, I.; Garikipati, A.; Xiong, X.; Nazo, N.; Rajagopal, S. β-Arrestin–Mediated Angiotensin II Type 1 Receptor Activation Promotes Pulmonary Vascular Remodeling in Pulmonary Hypertension. JACC Basic Transl. Sci. 2021, 6, 854–869. [Google Scholar] [CrossRef]
- Violin, J.D.; Lefkowitz, R.J. Beta-Arrestin-Biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 2007, 28, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; DeWire, S.M.; Yamashita, D.; Rominger, D.H.; Nguyen, L.; Schiller, K.; Whalen, E.J.; Gowen, M.; Lark, M.W. Selectively Engaging β-Arrestins at the Angiotensin II Type 1 Receptor Reduces Blood Pressure and Increases Cardiac Performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Bohn, L.M. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 2011, 60, 58–65. [Google Scholar] [CrossRef]
- Chiang, T.; Sansuk, K.; van Rijn, R.M. β-Arrestin 2 dependence of δ opioid receptor agonists is correlated with alcohol intake. Br. J. Pharmacol. 2016, 173, 332–343. [Google Scholar] [CrossRef]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175.e13. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.M.; Zell, V.; Faget, L.; Slosky, L.M.; Barak, L.S.; Caron, M.G.; Pinkerton, A.B.; Hnasko, T.S. Neurotensin receptor 1-biased ligand attenuates neurotensin-mediated excitation of ventral tegmental area dopamine neurons and dopamine release in the nucleus accumbens. Neuropharmacology 2023, 234, 109544. [Google Scholar] [CrossRef]
- Beom, S.; Cheong, D.; Torres, G.; Caron, M.G.; Kim, K.-M. Comparative Studies of Molecular Mechanisms of Dopamine D2 and D3 Receptors for the Activation of Extracellular Signal-regulated Kinase. J. Biol. Chem. 2004, 279, 28304–28314. [Google Scholar] [CrossRef] [PubMed]
- Kiss, B.; Laszlovszky, I.; Krámos, B.; Visegrády, A.; Bobok, A.; Lévay, G.; Lendvai, B.; Román, V. Neuronal Dopamine D3 Receptors: Translational Implications for Preclinical Research and CNS Disorders. Biomolecules 2021, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Hall, H.; Halldin, C.; Dijkstra, D.; Wikström, H.; Wise, L.D.; Pugsley, T.A.; Sokoloff, P.; Pauli, S.; Farde, L.; Sedvall, G. Autoradiographic localisation of D3-dopamine receptors in the human brain using the selective D3-dopamine receptor agonist (+)-[3H]PD 128907. Psychopharmacology 1996, 128, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Hurd, Y.L.; Sokoloff, P.; Schwartz, J.-C.; Sedvall, G. D3 dopamine receptor mRNA is widely expressed in the human brain. Brain Res. 1998, 779, 58–74. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Joyce, J.N. Distribution of Dopamine D3 Receptor Expressing Neurons in the Human Forebrain: Comparison with D2 Receptor Expressing Neurons. Neuropsychopharmacology 1999, 20, 60–80. [Google Scholar] [CrossRef]
- Nakajima, S.; Gerretsen, P.; Takeuchi, H.; Caravaggio, F.; Chow, T.; Le Foll, B.; Mulsant, B.; Pollock, B.; Graff-Guerrero, A. The potential role of dopamine D3 receptor neurotransmission in cognition. Eur. Neuropsychopharmacol. 2013, 23, 799–813. [Google Scholar] [CrossRef]
- Heidbreder, C.A.; Gardner, E.L.; Xi, Z.-X.; Thanos, P.K.; Mugnaini, M.; Hagan, J.J.; Ashby, C.R. The role of central dopamine D3 receptors in drug addiction: A review of pharmacological evidence. Brain Res. Rev. 2005, 49, 77–105. [Google Scholar] [CrossRef]
- Shafer, R.A.; Levant, B. The D3 dopamine receptor in cellular and organismal function. Psychopharmacology 1998, 135, 1–16. [Google Scholar] [CrossRef]
- Giros, B.; Martres, M.P.; Sokoloff, P.; Schwartz, J.C. Gene cloning of human dopaminergic D3 receptor and identification of its chromosome. Comptes Rendus Acad. Sci. III 1990, 311, 501–508. [Google Scholar]
- Westrich, L.; Kuzhikandathil, E.V. The tolerance property of human D3 dopamine receptor is determined by specific amino acid residues in the second cytoplasmic loop. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2007, 1773, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Kuzhikandathil, E.V.; Westrich, L.; Bakhos, S.; Pasuit, J. Identification and characterization of novel properties of the human D3 dopamine receptor. Mol. Cell. Neurosci. 2004, 26, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.W.; Jarvie, K.R.; Caron, M.G. High affinity agonist binding to the dopamine D3 receptor: Chimeric receptors delineate a role for intracellular domains. Mol. Pharmacol. 1994, 46, 352–356. [Google Scholar] [PubMed]
- Min, C.; Zheng, M.; Zhang, X.; Caron, M.G.; Kim, K.M. Novel roles for β-arrestins in the regulation of pharmacological sequestration to predict agonist-induced desensitization of dopamine D3 receptors. Br. J. Pharmacol. 2013, 170, 1112–1129. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Bearoff, F.; Kortagere, S. G-Protein biased signaling agonists of Dopamine D3 receptor promote distinct activation patterns of ERK1/2. Pharmacol. Res. 2022, 179, 106223. [Google Scholar] [CrossRef]
- Kim, K.-M.; Valenzano, K.J.; Robinson, S.R.; Yao, W.D.; Barak, L.S.; Caron, M.G. Differential Regulation of the Dopamine D2 and D3 Receptors by G Protein-coupled Receptor Kinases and β-Arrestins. J. Biol. Chem. 2001, 276, 37409–37414. [Google Scholar] [CrossRef]
- Sánchez-Soto, M.; Boldizsar, N.M.; Schardien, K.A.; Madaras, N.S.; Willette, B.K.A.; Inbody, L.R.; Dasaro, C.; Moritz, A.E.; Drube, J.; Haider, R.S.; et al. G Protein-Coupled Receptor Kinase 2 Selectively Enhances β-Arrestin Recruitment to the D2 Dopamine Receptor through Mechanisms That Are Independent of Receptor Phosphorylation. Biomolecules 2023, 13, 1552. [Google Scholar] [CrossRef]
- Westrich, L.; Gil-Mast, S.; Kortagere, S.; Kuzhikandathil, E.V. Development of tolerance in D3 dopamine receptor signaling is accompanied by distinct changes in receptor conformation. Biochem. Pharmacol. 2010, 79, 897–907. [Google Scholar] [CrossRef]
- Gil-Mast, S.; Kortagere, S.; Kota, K.; Kuzhikandathil, E.V. An Amino Acid Residue in the Second Extracellular Loop Determines the Agonist-Dependent Tolerance Property of the Human D3 Dopamine Receptor. ACS Chem. Neurosci. 2013, 4, 940–951. [Google Scholar] [CrossRef]
- Schneider, J.S.; Marshall, C.A.; Keibel, L.; Snyder, N.W.; Hill, M.P.; Brotchie, J.M.; Johnston, T.H.; Waterhouse, B.D.; Kortagere, S. A novel dopamine D3R agonist SK609 with norepinephrine transporter inhibition promotes improvement in cognitive task performance in rodent and non-human primate models of Parkinson’s disease. Exp. Neurol. 2021, 335, 113514. [Google Scholar] [CrossRef]
- Simms, S.L.; Huettner, D.P.; Kortagere, S. In vivo characterization of a novel dopamine D3 receptor agonist to treat motor symptoms of Parkinson’s disease. Neuropharmacology 2016, 100, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Reith, M.E.A.; Liu-Chen, L.-Y.; Kortagere, S. Biased signaling agonist of dopamine D3 receptor induces receptor internalization independent of β-arrestin recruitment. Pharmacol. Res. 2019, 143, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Free, R.B.; Chun, L.S.; Moritz, A.E.; Miller, B.N.; Doyle, T.B.; Conroy, J.L.; Padron, A.; Meade, J.A.; Xiao, J.; Hu, X.; et al. Discovery and characterization of a G protein-biased agonist that inhibits β-arrestin recruitment to the D2 dopamine receptor. Mol. Pharmacol. 2014, 86, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.M.; Pack, T.F.; Wilkins, A.D.; Urs, N.M.; Urban, D.J.; Bass, C.E.; Lichtarge, O.; Caron, M.G. Elucidation of G-protein and β-arrestin functional selectivity at the dopamine D2 receptor. Proc. Natl. Acad. Sci. USA 2015, 112, 7097–7102. [Google Scholar] [CrossRef]
- Cahill, T.J.; Thomsen, A.R.B.; Tarrasch, J.T.; Plouffe, B.; Nguyen, A.H.; Yang, F.; Huang, L.; Kahsai, A.W.; Bassoni, D.L.; Gavino, B.J.; et al. Distinct conformations of GPCR–β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. USA 2017, 114, 2562–2567. [Google Scholar] [CrossRef]
- Chaturvedi, M.; Maharana, J.; Shukla, A.K. Terminating G-Protein Coupling: Structural Snapshots of GPCR-β-Arrestin Complexes. Cell 2020, 180, 1041–1043. [Google Scholar] [CrossRef]
- Oakley, R.H.; Olivares-Reyes, J.A.; Hudson, C.C.; Flores-Vega, F.; Dautzenberg, F.M.; Hauger, R.L. Carboxyl-terminal and intracellular loop sites for CRF1 receptor phosphorylation and β-arrestin-2 recruitment: A mechanism regulating stress and anxiety responses. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 293, R209–R222. [Google Scholar] [CrossRef]
- Verweij, E.W.E.; Araaj, B.A.; Prabhata, W.R.; Prihandoko, R.; Nijmeijer, S.; Tobin, A.B.; Leurs, R.; Vischer, H.F. Differential Role of Serines and Threonines in Intracellular Loop 3 and C-Terminal Tail of the Histamine H4 Receptor in β-Arrestin and G Protein-Coupled Receptor Kinase Interaction, Internalization, and Signaling. ACS Pharmacol. Transl. Sci. 2020, 3, 321–333. [Google Scholar] [CrossRef]
- Shukla, A.K.; Manglik, A.; Kruse, A.C.; Xiao, K.; Reis, R.I.; Tseng, W.-C.; Staus, D.P.; Hilger, D.; Uysal, S.; Huang, L.-Y.; et al. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 2013, 497, 137–141. [Google Scholar] [CrossRef]
- Lan, H.; Liu, Y.; Bell, M.I.; Gurevich, V.V.; Neve, K.A. A Dopamine D2 Receptor Mutant Capable of G Protein-Mediated Signaling but Deficient in Arrestin Binding. Mol. Pharmacol. 2009, 75, 113–123. [Google Scholar] [CrossRef]
- Kim, K.-M. Unveiling the Differences in Signaling and Regulatory Mechanisms between Dopamine D2 and D3 Receptors and Their Impact on Behavioral Sensitization. Int. J. Mol. Sci. 2023, 24, 6742. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.-Y.; Cho, D.-I.; Park, J.H.; Kurose, H.; Caron, M.G.; Kim, K.-M. Roles of Protein Kinase C and Actin-Binding Protein 280 in the Regulation of Intracellular Trafficking of Dopamine D3 Receptor. Mol. Endocrinol. 2007, 21, 2242–2254. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Huang, S.; Mao, C.; Krumm, B.E.; Zhou, X.E.; Tan, Y.; Huang, X.-P.; Liu, Y.; Shen, D.-D.; Jiang, Y.; et al. Structures of the human dopamine D3 receptor-Gi complexes. Mol. Cell 2021, 81, 1147–1159.e4. [Google Scholar] [CrossRef] [PubMed]
- Urs, N.M.; Peterson, S.M.; Caron, M.G. New Concepts in Dopamine D2 Receptor Biased Signaling and Implications for Schizophrenia Therapy. Biol. Psychiatry 2017, 81, 78–85. [Google Scholar] [CrossRef]
- Männel, B.; Hübner, H.; Möller, D.; Gmeiner, P. β-Arrestin biased dopamine D2 receptor partial agonists: Synthesis and pharmacological evaluation. Bioorg. Med. Chem. 2017, 25, 5613–5628. [Google Scholar] [CrossRef]
- Bonifazi, A.; Yano, H.; Guerrero, A.M.; Kumar, V.; Hoffman, A.F.; Lupica, C.R.; Shi, L.; Newman, A.H. Novel and Potent Dopamine D2 Receptor Go-Protein Biased Agonists. ACS Pharmacol. Transl. Sci. 2019, 2, 52–65. [Google Scholar] [CrossRef]
- Xu, W.; Wang, X.; Tocker, A.M.; Huang, P.; Reith, M.E.A.; Liu-Chen, L.-Y.; Smith, A.B.; Kortagere, S. Functional Characterization of a Novel Series of Biased Signaling Dopamine D3 Receptor Agonists. ACS Chem. Neurosci. 2017, 8, 486–500. [Google Scholar] [CrossRef]
- Asher, W.B.; Terry, D.S.; Gregorio, G.G.A.; Kahsai, A.W.; Borgia, A.; Xie, B.; Modak, A.; Zhu, Y.; Jang, W.; Govindaraju, A.; et al. GPCR-Mediated β-Arrestin activation deconvoluted with single-molecule precision. Cell 2022, 185, 1661–1675.e16. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Mao, L.-M.; Zhang, G.-C.; Papasian, C.J.; Fibuch, E.E.; Lan, H.-X.; Zhou, H.-F.; Xu, M.; Wang, J.Q. Activity-Dependent Modulation of Limbic Dopamine D3 Receptors by CaMKII. Neuron 2009, 61, 425–438. [Google Scholar] [CrossRef]
- McCorvy, J.D.; Butler, K.V.; Kelly, B.; Rechsteiner, K.; Karpiak, J.; Betz, R.M.; Kormos, B.L.; Shoichet, B.K.; O Dror, R.; Jin, J.; et al. Structure-inspired design of β-Arrestin-Biased ligands for aminergic GPCRs. Nat. Chem. Biol. 2018, 14, 126–134. [Google Scholar] [CrossRef]
- Uprety, R.; Che, T.; A Zaidi, S.; Grinnell, S.G.; Varga, B.R.; Faouzi, A.; Slocum, S.T.; Allaoa, A.; Varadi, A.; Nelson, M.; et al. Controlling opioid receptor functional selectivity by targeting distinct subpockets of the orthosteric site. eLife 2021, 10, e56519. [Google Scholar] [CrossRef] [PubMed]
- El Daibani, A.; Paggi, J.M.; Kim, K.; Laloudakis, Y.D.; Popov, P.; Bernhard, S.M.; Krumm, B.E.; Olsen, R.H.J.; Diberto, J.; Carroll, F.I.; et al. Molecular mechanism of biased signaling at the kappa opioid receptor. Nat. Commun. 2023, 14, 1338. [Google Scholar] [CrossRef] [PubMed]
- Suomivuori, C.-M.; Latorraca, N.R.; Wingler, L.M.; Eismann, S.; King, M.C.; Kleinhenz, A.L.W.; Skiba, M.A.; Staus, D.P.; Kruse, A.C.; Lefkowitz, R.J.; et al. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science 2020, 367, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Cong, X.; Maurel, D.; Déméné, H.; Vasiliauskaité-Brooks, I.; Hagelberger, J.; Peysson, F.; Saint-Paul, J.; Golebiowski, J.; Granier, S.; Sounier, R. Molecular insights into the biased signaling mechanism of the μ-opioid receptor. Mol. Cell 2021, 81, 4165–4175.e6. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, M.; Cho, A.; Ma, N.; Mukhaleva, E.; Namkung, Y.; Lee, S.; Ghosh, S.; Lee, J.H.; Gloriam, D.E.; Laporte, S.A.; et al. Dynamic spatiotemporal determinants modulate GPCR:G protein coupling selectivity and promiscuity. Nat. Commun. 2022, 13, 7428. [Google Scholar] [CrossRef]
- Yin, W.; Li, Z.; Jin, M.; Yin, Y.-L.; de Waal, P.W.; Pal, K.; Yin, Y.; Gao, X.; He, Y.; Gao, J.; et al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 2019, 29, 971–983. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Tukey, J.W. Comparing Individual Means in the Analysis of Variance. Biometrics 1949, 5, 99–114. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g: Profiler—Interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2022, 51, D587–D592. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2021, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Balcı, H.; Hanspers, K.; Coort, S.L.; Martens, M.; Slenter, D.N.; Ehrhart, F.; Digles, D.; Waagmeester, A.; Wassink, I.; et al. WikiPathways 2024: Next generation pathway database. Nucleic Acids Res. 2023, 52, D679–D689. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; A Craig, P.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2022, 51, D488–D508. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.; Ohnmacht, U.; Lehmann, T.; Utz, W.; Hübner, H.; Gmeiner, P. CoMFA and CoMSIA Investigations Revealing Novel Insights into the Binding Modes of Dopamine D3 Receptor Agonists. J. Med. Chem. 2005, 48, 2493–2508. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2022.02; Chemical Computing Group ULC: Montreal, QC, Canada, 2023.
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis SC’ 14, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Cao, C.; Barros-Álvarez, X.; Zhang, S.; Kim, K.; Dämgen, M.A.; Panova, O.; Suomivuori, C.-M.; Fay, J.F.; Zhong, X.; Krumm, B.E.; et al. Signaling snapshots of a serotonin receptor activated by the prototypical psychedelic LSD. Neuron 2022, 110, 3154–3167.e7. [Google Scholar] [CrossRef]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-Arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef]
- Bous, J.; Fouillen, A.; Orcel, H.; Trapani, S.; Cong, X.; Fontanel, S.; Saint-Paul, J.; Lai-Kee-Him, J.; Urbach, S.; Sibille, N.; et al. Structure of the vasopressin hormone–V2 receptor–β-Arrestin1 ternary complex. Sci. Adv. 2022, 8, eabo7761. [Google Scholar] [CrossRef]
- Szczepek, M.; Beyrière, F.; Hofmann, K.P.; Elgeti, M.; Kazmin, R.; Rose, A.; Bartl, F.J.; von Stetten, D.; Heck, M.; Sommer, M.E.; et al. Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat. Commun. 2014, 5, 4801. [Google Scholar] [CrossRef]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef]
- Sente, A.; Peer, R.; Srivastava, A.; Baidya, M.; Lesk, A.M.; Balaji, S.; Shukla, A.K.; Babu, M.M.; Flock, T. Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol. 2018, 25, 538–545. [Google Scholar] [CrossRef]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-Arrestin coupling to formoterol-bound β1-Adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Yoon, H.-J.; Park, J.Y.; Baidya, M.; Dwivedi-Agnihotri, H.; Maharana, J.; Chaturvedi, M.; Chung, K.Y.; Shukla, A.K.; Lee, H.H. Crystal Structure of β-Arrestin 2 in Complex with CXCR7 Phosphopeptide. Structure 2020, 28, 1014–1023.e4. [Google Scholar] [CrossRef] [PubMed]
- Maharana, J.; Sarma, P.; Yadav, M.K.; Saha, S.; Singh, V.; Saha, S.; Chami, M.; Banerjee, R.; Shukla, A.K. Structural snapshots uncover a key phosphorylation motif in GPCRs driving β-Arrestin activation. Mol. Cell 2023, 83, 2091–2107.e7. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | TM3 | TM5 | TM6 | TM7 | ILE183 |
|---|---|---|---|---|---|
| D3R-PRX | −55.78 ± 5.5 | −6.8 ± 1.8 | 4.3 ± 18.6 | 2.5 ± 12.3 | 2.0 ± 9.3 |
| D3R-PRX-Gi | −45.31 ± 9.8 | −6.4 ± 2.7 | −1.6 ± 24.9 | 5.5 ± 8.0 | 0.3 ± 3.6 |
| D3R-βarr2-a-SK609 | −48.1 ± 8.8 | −6.4 ± 2.2 | −0.8 ± 8.9 | 2.3 ± 10.1 | 3.1 ± 3.1 |
| D3R-βarr2-b-PRX | −29.6 ± 17.0 | −10.1 ± 2.5 | −9.3 ± 14.9 | 1.2 ± 0.5 | 2.2 ± 6.0 |
| D3R- SK609 | −66.2 ± 6.3 | −5.0 ± 1.1 | −3.4 ± 5.1 | −1.2 ± 2.5 | −0.3 ± 1.6 |
| D3R- SK609-Gi | −66.7 ± 7.6 | −5.3 ± 1.1 | −7.2 ± 10.7 | 0.6 ± 2.2 | −0.4 ± 3.3 |
| D3R-βarr2-a-SK609 | −68.6 ± 4.1 | −5.5 ± 0.8 | −1.7 ± 5.2 | −1.4 ± 1.4 | −0.2 ± 1.5 |
| D3R-βarr2-b-SK609 | −65.7 ± 8.3 | −5.7 ± 1.7 | −4.6 ± 4.7 | −0.3 ± 2.5 | −0.5 ± 1.8 |
| Complex | TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 |
|---|---|---|---|---|---|---|---|
| D3R-PRX | 27 ± 3 | 29 ± 3 | 30 ± 3 | 6 ± 3 | 34 ± 3 | 7 ± 3 | 18 ± 4 |
| D3R-SK609 | 22 ± 3 | 27 ± 3 | 25 ± 3 | 7 ± 3 | 32 ± 3 | 12 ± 4 | 23 ± 4 |
| D3R(Leu121Asn, Tyr129Leu)-PRX | 25 ± 3 | 29 ± 3 | 24 ± 3 | 6 ± 3 | 32 ± 3 | 17 ± 5 | 20 ± 4 |
| D3R-βarr2-b-PRX | 32 ± 3 | 32 ± 2 | 30 ± 2 | 13 ± 3 | 28 ± 3 | 7 ± 3 | 25 ± 4 |
| D3R-βarr2-b-SK609 | 29 ± 3 | 30 ± 2 | 30 ± 2 | 7 ± 2 | 31 ± 3 | 8 ± 3 | 31 ± 3 |
| D3R-Gi-PRX | 32 ± 5 | 24 ± 3 | 17 ± 3 | 17 ± 3 | 12 ± 4 | 7 ± 3 | 11 ± 4 |
| D3R-Gi-SK609 | 35 ± 4 | 26 ± 3 | 19 ± 2 | 13 ± 4 | 21 ± 4 | 11 ± 3 | 10 ± 7 |
| Ligand | D3R_βarr2 | TM2_fl | TM3_fl | TM6_fl | TM7_fl | ICL1_ βarr2 | ICL2_ βarr2 | ICL3_ βarr2 |
|---|---|---|---|---|---|---|---|---|
| D3R-βarr2-a complexes | ||||||||
| PRX | −296.8 ± 51.1 | −5.4 ± 6.2 | −24.5 ± 10.5 | −20.4 ± 7.3 | −1.4 ± 1.31 | 1.87 ± 11.6 | −50.7 ± 11.6 | −163.1 ± 42.2 |
| SK609 | −364.2 ± 41.2 | −0.2 ± 0.6 | −78.2 ± 6.5 | −9.3 ± 5.8 | −0.3 ± 0.7 | 12.9 ± 10.3 | −89.7 ± 12.4 | −146.7 ± 29.7 |
| D3R-βarr2-b complexes | ||||||||
| PRX | −596.1 ± 70.1 | −54.2 ± 7.8 | −67.4 ± 10.4 | −22.6 ± 5.3 | −9.3 ± 2.5 | −46.8 ± 14.0 | −97.6 ± 12.7 | −247.4 ± 55.0 |
| SK609 | −480.2 ± 52.1 | −17.9 ± 6.2 | −63.5 ± 2.9 | −17.6 ± 3.2 | −6.2 ± 1.8 | −8.1 ± 9.6 | −113.6 ± 19.4 | −214.4 ± 43.1 |
| Complex | Protein–Ligand | Protein–Protein |
|---|---|---|
| D3R-PRX | −25.03 ± 2.63 | |
| D3R-SK609 | −41.65 ± 4.60 | |
| D3R-βarr2-a-PRX | −28.88 ± 4.06 | 65.23 ± 18.77 |
| D3R-βarr2-a-SK609 | −40.86 ± 3.68 | 10.35 ± 24.38 |
| D3R-βarr2-b-PRX | −21.15 ± 3.91 | −273.93 ± 27.52 |
| D3R-βarr2-b-SK609 | −38.90 ± 3.55 | −198.50 ± 18.95 |
| D3R-Gi-PRX | −20.36 ± 15.11 | −287.33 ± 24.29 |
| D3R-Gi-SK609 | −36.17 ± 2.70 | −385.08 ± 35.78 |
| Complex | D187_H354 | D127_T63 | D127_T64 |
|---|---|---|---|
| D3R-PRX | 7.2 ± 1.7 | 4.9 ± 1.6 | 1.9 ± 0.8 |
| D3R-SK609 | 10.3 ± 2.0 | 1.8 ± 0.2 | 1.9 ± 0.7 |
| D3R(Leu121Asn, Tyr129Leu)-PRX | 10.3 ± 1.7 | 1.8 ± 0.3 | 1.7 ± 0.2 |
| D3R-βarr2-b-PRX | 13.9 ± 0.7 | 2.1 ± 1.0 | 5.8 ± 1.2 |
| D3R-βarr2-b-SK609 | 14.6 ± 1.3 | 8.9 ± 0.6 | 6.3 ± 0.8 |
| D3R-Gi-PRX | 9.3 ± 1.6 | 1.8 ± 0.3 | 2.4 ± 1.2 |
| D3R-Gi-SK609 | 9.1 ± 1.8 | 3.0 ± 1.5 | 2.2 ± 1.0 |
| Complex | TM3-TM7 | TM2-TM6 |
|---|---|---|
| D3R-PRX | 18.7 ± 0.9 | 17.5 ± 0.9 |
| D3R-SK609 | 23.6 ± 1.0 | 16.7 ± 0.8 |
| D3R(Leu121Asn, Tyr129Leu)-PRX | 22.9 ± 0.9 | 19.7 ± 0.8 |
| D3R-βarr2-b-PRX | 27.1 ± 0.6 | 18.6 ± 0.7 |
| D3R-βarr2-b-SK609 | 27.1 ± 0.6 | 19.4 ± 0.6 |
| D3R-PRX-Gi | 25.7 ± 1.0 | 18.8 ± 0.5 |
| D3R-SK609-Gi | 23.7 ± 1.1 | 20.4 ± 0.5 |
| Systems | Number of Lipids | Patch Dimension Å × Å (Å2) | Simulation Time |
|---|---|---|---|
| 131 | 73 × 73 | 1 µs |
| 131 | 73 × 73 | 1 µs |
| 133 | 73 × 73 | 1 µs |
| 327 | 105 × 105 | 1 µs |
| 331 | 105 × 105 | 1 µs |
| 323 | 103 × 103 | 1 µs |
| 333 | 103 × 103 | 1 µs |
| 301 | 101 × 101 | 2 µs |
| 301 | 101 × 101 | 2 µs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nepal, B.; Barnett, J.; Bearoff, F.; Kortagere, S. Biased Signaling Agonists Promote Distinct Phosphorylation and Conformational States of the Dopamine D3 Receptor. Int. J. Mol. Sci. 2024, 25, 10470. https://doi.org/10.3390/ijms251910470
Nepal B, Barnett J, Bearoff F, Kortagere S. Biased Signaling Agonists Promote Distinct Phosphorylation and Conformational States of the Dopamine D3 Receptor. International Journal of Molecular Sciences. 2024; 25(19):10470. https://doi.org/10.3390/ijms251910470
Chicago/Turabian StyleNepal, Binod, Jessica Barnett, Frank Bearoff, and Sandhya Kortagere. 2024. "Biased Signaling Agonists Promote Distinct Phosphorylation and Conformational States of the Dopamine D3 Receptor" International Journal of Molecular Sciences 25, no. 19: 10470. https://doi.org/10.3390/ijms251910470