
Characterization of Endothelial Cell Subclusters in Localized Scleroderma Skin with Single-Cell RNA Sequencing Identifies NOTCH Signaling Pathway

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
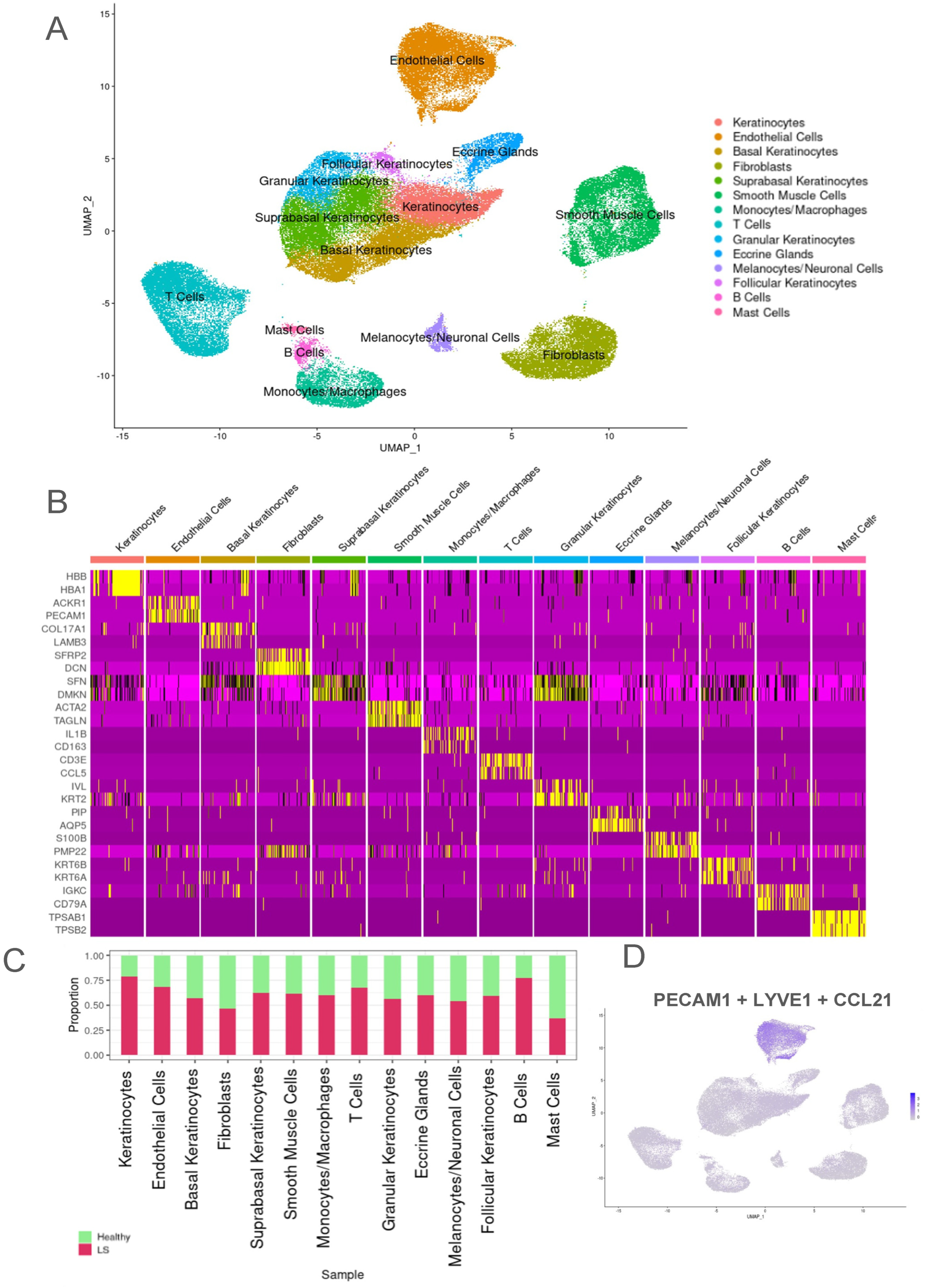
2.1. Transcriptomic Evaluation of Endothelial Cells from Both LS and Healthy Control Samples Identified Higher Proportion of LS Samples in the Endothelial Cell Population
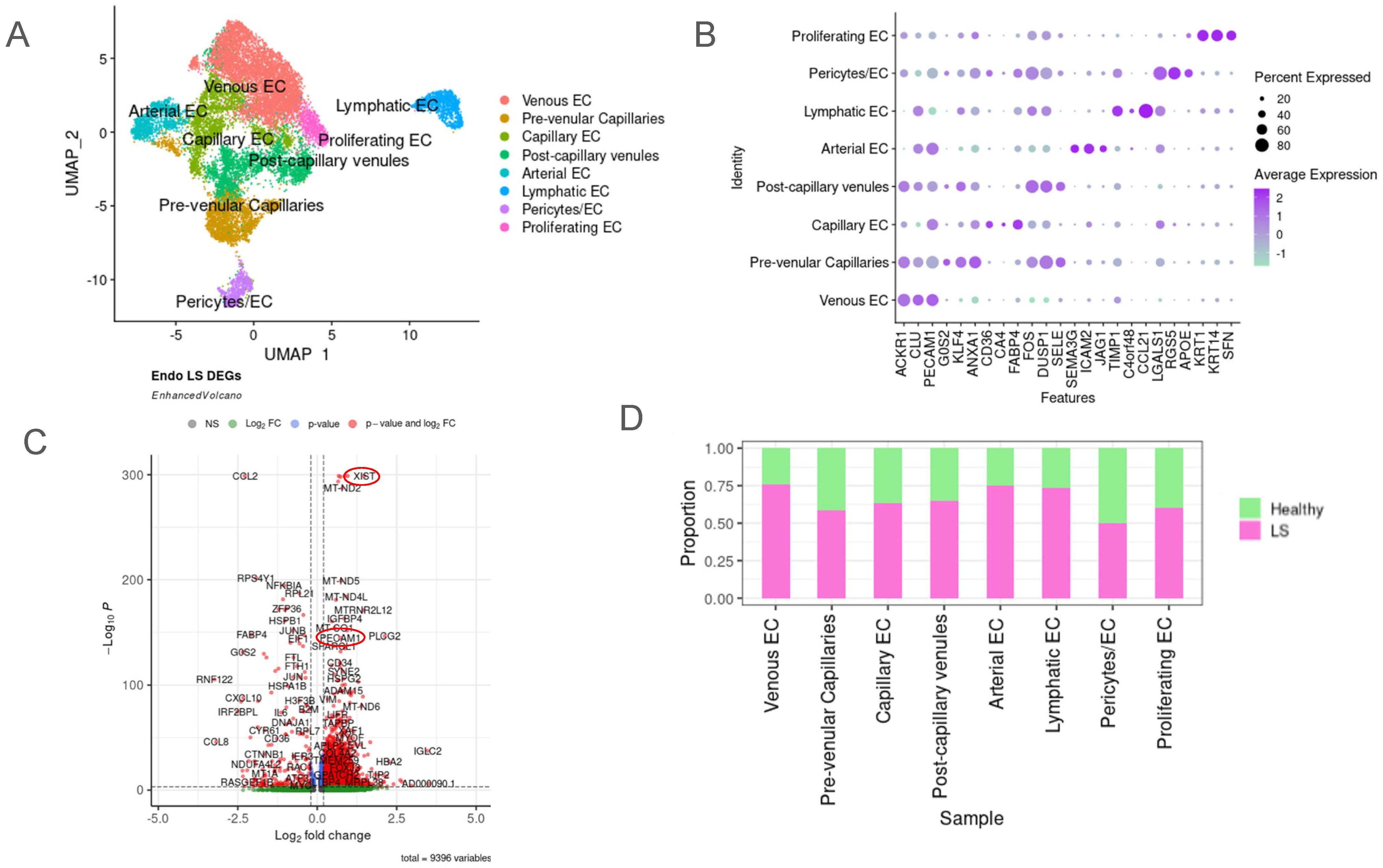
2.2. Unique Subclusters of Endothelial Cells in LS Were Identified and Their Differentially Expressed Genes Were Significant When Compared with Healthy Controls
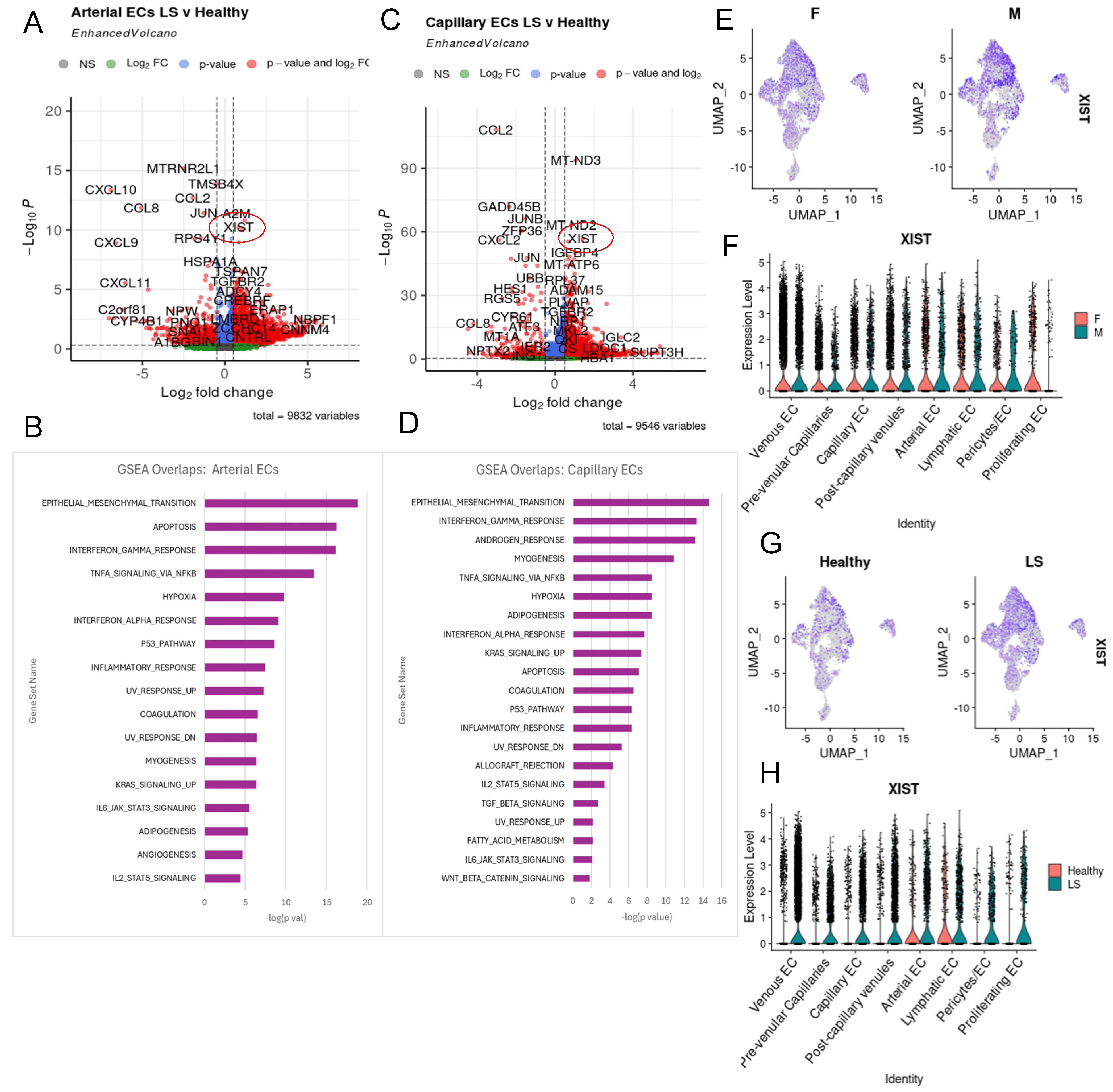
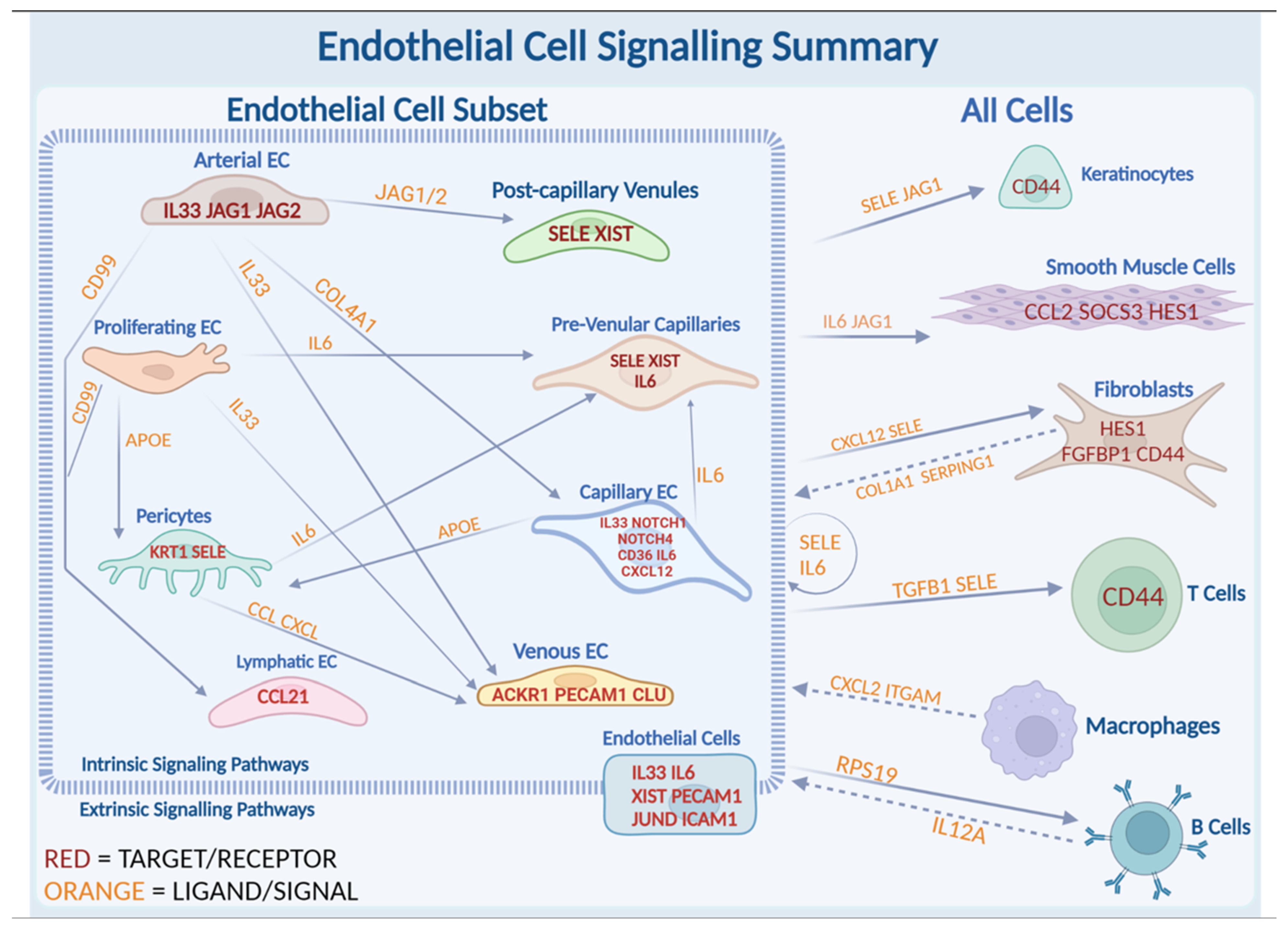
2.3. JAG/NOTCH Signaling between Arterial Endothelial Cells and Capillary Endothelial Cells
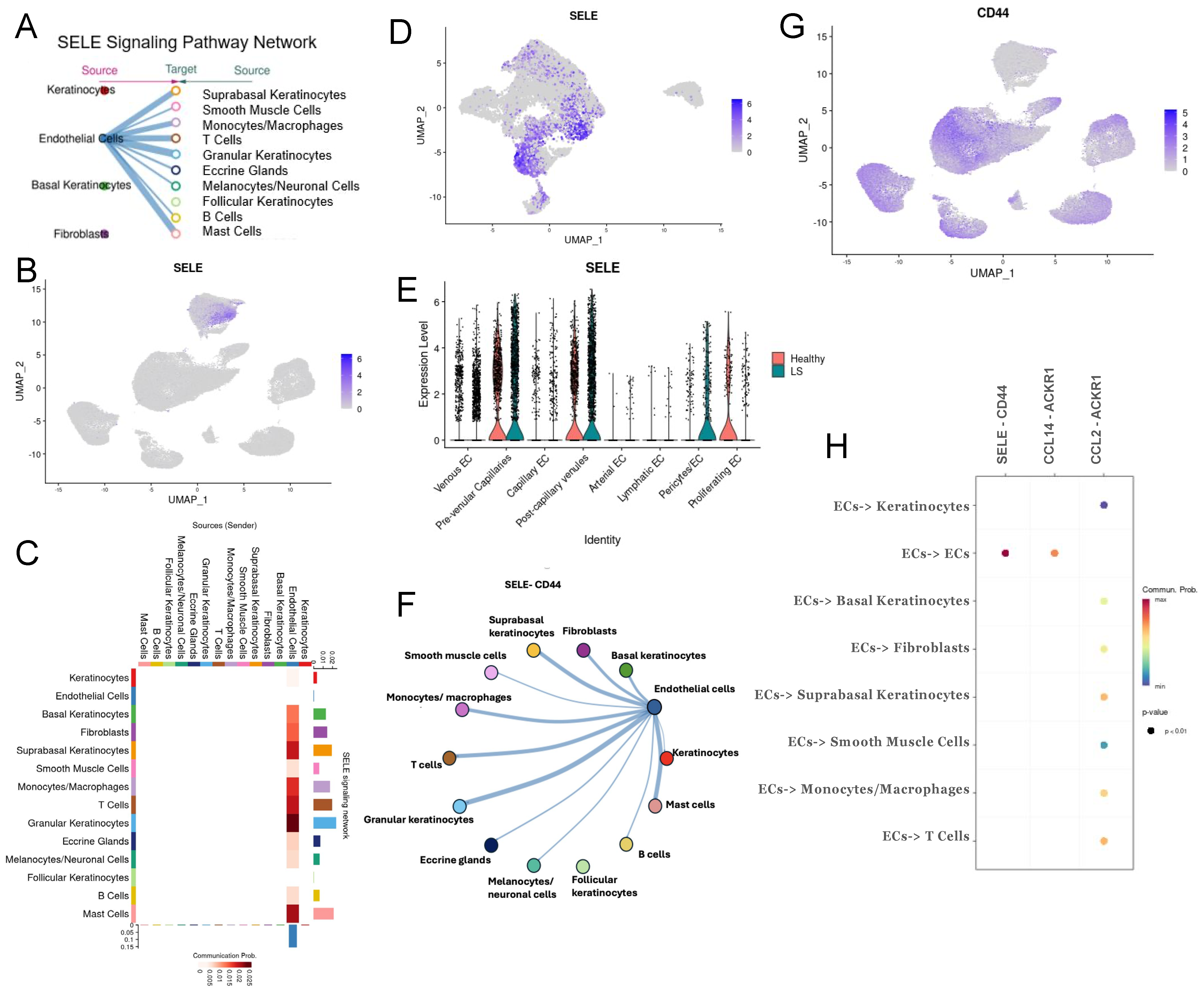
2.4. Extrinsic SELE Signaling Originating from Endothelial Cells with Predicted Receptor CD44 and Extracellular Communications and Crosstalk between JAG/NOTCH Pathway
2.5. XIST Expression Higher in LS across All Endothelial Cell Subtypes
2.6. Potential Roles of IL-33 and IL6 in LS Endothelial Cells
3. Discussion
Limitations
4. Materials and Methods
4.1. Human-Patient Skin Samples
4.2. Single-Cell RNA Sequencing
4.3. Spatial Transcriptomics
4.4. Data Preprocessing and Bioinformatics Analyses
4.5. Cell-Communication Analysis via NicheNet and CellChat
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, S.C. Scleroderma in Children and Adolescents: Localized Scleroderma and Systemic Sclerosis. Pediatr. Clin. N. Am. 2018, 65, 757–781. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Rosa, I.; Fioretto, B.S.; Matucci-Cerinic, M.; Manetti, M. New Insights into Profibrotic Myofibroblast Formation in Systemic Sclerosis: When the Vascular Wall Becomes the Enemy. Life 2021, 11, 610. [Google Scholar] [CrossRef]
- Kurzinski, K.L.; Zigler, C.K.; Torok, K.S. Prediction of disease relapse in a cohort of paediatric patients with localized scleroderma. Br. J. Dermatol. 2019, 180, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.M.; LeRoy, E.C. Pathogenesis of systemic sclerosis: A vascular hypothesis. Semin. Arthritis Rheumatol. 1975, 4, 351–368. [Google Scholar] [CrossRef]
- Prescott, R.J.; Freemont, A.J.; Jones, C.J.; Hoyland, J.; Fielding, P. Sequential dermal microvascular and perivascular changes in the development of scleroderma. J. Pathol. 1992, 166, 255–263. [Google Scholar] [CrossRef]
- Freemont, A.J.; Jones, C.J.; Bromley, M.; Andrews, P. Changes in vascular endothelium related to lymphocyte collections in diseased synovia. Arthritis Rheumatol. 1983, 26, 1427–1433. [Google Scholar] [CrossRef]
- Fleischmajer, R.; Perlish, J.S.; Shaw, K.V.; Pirozzi, D.J. Skin capillary changes in early systemic scleroderma. Electron microscopy and “in vitro” autoradiography with tritiated thymidine. Arch. Dermatol. 1976, 112, 1553–1557. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.; Susa, J.S.; Currimbhoy, S.; Jacobe, H. Histopathological changes in morphea and their clinical correlates: Results from the Morphea in Adults and Children Cohort V. J. Am. Acad. Dermatol. 2017, 76, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Roumm, A.D.; Whiteside, T.L.; Medsger, T.A., Jr.; Rodnan, G.P. Lymphocytes in the skin of patients with progressive systemic sclerosis. Quantification, subtyping, and clinical correlations. Arthritis Rheumatol. 1984, 27, 645–653. [Google Scholar] [CrossRef]
- Schachna, L.; Wigley, F.M. Targeting mediators of vascular injury in scleroderma. Curr. Opin. Rheumatol. 2002, 14, 686–693. [Google Scholar] [CrossRef]
- Manetti, M.; Guiducci, S.; Romano, E.; Rosa, I.; Ceccarelli, C.; Mello, T.; Milia, A.F.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Differential expression of junctional adhesion molecules in different stages of systemic sclerosis. Arthritis Rheumatol. 2013, 65, 247–257. [Google Scholar] [CrossRef]
- Lunardi, C.; Dolcino, M.; Peterlana, D.; Bason, C.; Navone, R.; Tamassia, N.; Beri, R.; Corrocher, R.; Puccetti, A. Antibodies against human cytomegalovirus in the pathogenesis of systemic sclerosis: A gene array approach. PLoS Med. 2006, 3, e2. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Okazaki, Y. Quantification of circulating endothelial progenitor cells in systemic sclerosis: A direct comparison of protocols. Ann. Rheum. Dis. 2012, 71, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Del Papa, N.; Colombo, G.; Fracchiolla, N.; Moronetti, L.M.; Ingegnoli, F.; Maglione, W.; Comina, D.P.; Vitali, C.; Fantini, F.; Cortelezzi, A. Circulating endothelial cells as a marker of ongoing vascular disease in systemic sclerosis. Arthritis Rheumatol. 2004, 50, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D.; Illingworth, K.; Jayson, M.I. Mechanisms of endothelial cell damage in systemic sclerosis and Raynaud’s phenomenon. J. Rheumatol. 1993, 20, 1325–1330. [Google Scholar]
- Kurzinski, K.; Torok, K.S. Cytokine profiles in localized scleroderma and relationship to clinical features. Cytokine 2011, 55, 157–164. [Google Scholar] [CrossRef]
- Rice, L.M.; Ziemek, J.; Stratton, E.A.; McLaughlin, S.R.; Padilla, C.M.; Mathes, A.L.; Christmann, R.B.; Stifano, G.; Browning, J.L.; Whitfield, M.L.; et al. A longitudinal biomarker for the extent of skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 2015, 67, 3004–3015. [Google Scholar] [CrossRef]
- Jun, J.-B.; Kuechle, M.; Harlan, J.M.; Elkon, K.B. Fibroblast and endothelial apoptosis in systemic sclerosis. Curr. Opin. Rheumatol. 2003, 15, 756–760. [Google Scholar] [CrossRef]
- Apostolidis, S.A.; Stifano, G.; Tabib, T.; Rice, L.M.; Morse, C.M.; Kahaleh, B.; Lafyatis, R. Single Cell RNA Sequencing Identifies HSPG2 and APLNR as Markers of Endothelial Cell Injury in Systemic Sclerosis Skin. Front. Immunol. 2018, 9, 2191. [Google Scholar] [CrossRef] [PubMed]
- Matucci-Cerinic, M.; Kahaleh, B.; Wigley, F.M. Review: Evidence that systemic sclerosis is a vascular disease. Arthritis Rheumatol. 2013, 65, 1953–1962. [Google Scholar] [CrossRef]
- Choi, Y.-S.; Choi, H.-J.; Min, J.-K.; Pyun, B.-J.; Maeng, Y.-S.; Park, H.; Kim, J.; Kim, Y.-M.; Kwon, Y.-G. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood 2009, 114, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Mostmans, Y.; Cutolo, M.; Giddelo, C.; Decuman, S.; Melsens, K.; Declercq, H.; Vandecasteele, E.; De Keyser, F.; Distler, O.; Gutermuth, J.; et al. The role of endothelial cells in the vasculopathy of systemic sclerosis: A systematic review. Autoimmun. Rev. 2017, 16, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Vettori, S.; Cuomo, G.; Iudici, M.; D’Abrosca, V.; Giacco, V.; Barra, G.; De Palma, R.; Valentini, G. Early systemic sclerosis: Serum profiling of factors involved in endothelial, T-cell, and fibroblast interplay is marked by elevated interleukin-33 levels. J. Clin. Immunol. 2014, 34, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Guiducci, S.; Ceccarelli, C.; Romano, E.; Bellando-Randone, S.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Increased circulating levels of interleukin 33 in systemic sclerosis correlate with early disease stage and microvascular involvement. Ann. Rheum. Dis. 2011, 70, 1876–1878. [Google Scholar] [CrossRef] [PubMed]
- Terras, S.; Opitz, E.; Moritz, R.K.C.; Höxtermann, S.; Gambichler, T.; Kreuter, A. Increased serum IL-33 levels may indicate vascular involvement in systemic sclerosis. Ann. Rheum. Dis. 2013, 72, 144–145. [Google Scholar] [CrossRef]
- Song, M.; Wang, Y.; Annex, B.H.; Popel, A.S. Experiment-based Computational Model Predicts that IL-6 Trans-Signaling Plays a Dominant Role in IL-6 mediated signaling in Endothelial Cells. bioRxiv 2023. [Google Scholar] [CrossRef]
- Signorelli, S.S.; Fiore, V.; Malaponte, G. Inflammation and peripheral arterial disease: The value of circulating biomarkers (Review). Int. J. Mol. Med. 2014, 33, 777–783. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Mirizio, E.; Tabib, T.; Wang, X.; Chen, W.; Liu, C.; Lafyatis, R.; Jacobe, H.; Torok, K.S. Single-cell transcriptome conservation in a comparative analysis of fresh and cryopreserved human skin tissue: Pilot in localized scleroderma. Arthritis Res. Ther. 2020, 22, 263. [Google Scholar] [CrossRef]
- Werner, G.; Sanyal, A.; Mirizio, E.; Hutchins, T.; Tabib, T.; Lafyatis, R.; Jacobe, H.; Torok, K.S. Single-Cell Transcriptome Analysis Identifies Subclusters with Inflammatory Fibroblast Responses in Localized Scleroderma. Int. J. Mol. Sci. 2023, 24, 9796. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.G.; Zigmond, Z.M.; Pereira-Simon, S.; Falcon, N.S.; Kumar, M.S.; Stoyell-Conti, F.F.; Kosanovic, C.; Griswold, A.J.; Salama, A.; Yang, X.; et al. The intricate cellular ecosystem of human peripheral veins as revealed by single-cell transcriptomic analysis. PLoS ONE 2024, 19, e0296264. [Google Scholar] [CrossRef] [PubMed]
- Vaahtomeri, K.; Moussion, C.; Hauschild, R.; Sixt, M. Shape and Function of Interstitial Chemokine CCL21 Gradients Are Independent of Heparan Sulfates Produced by Lymphatic Endothelium. Front. Immunol. 2021, 12, 630002. [Google Scholar] [CrossRef] [PubMed]
- Denzer, L.; Muranyi, W.; Schroten, H.; Schwerk, C. The role of PLVAP in endothelial cells. Cell Tissue Res. 2023, 392, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.K.; Wilson, C.H.; Harding, K.G.; Finlay, A.Y.; Bowden, P.E. Numerous keratinocyte subtypes involved in wound re-epithelialization. J. Investig. Dermatol. 2006, 126, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Cai, L.; Cui, C.; de Los Toyos, J.R.; Anastassiou, D. Single-cell analysis reveals the pan-cancer invasiveness-associated transition of adipose-derived stromal cells into COL11A1-expressing cancer-associated fibroblasts. PLoS Comput. Biol. 2021, 17, e1009228. [Google Scholar] [CrossRef]
- Theocharidis, G.; Thomas, B.E.; Sarkar, D.; Mumme, H.L.; Pilcher, W.J.R.; Dwivedi, B.; Sandoval-Schaefer, T.; Sîrbulescu, R.F.; Kafanas, A.; Mezghani, I.; et al. Single cell transcriptomic landscape of diabetic foot ulcers. Nat. Commun. 2022, 13, 181. [Google Scholar] [CrossRef]
- Sun, J.; Qiao, Y.-N.; Tao, T.; Zhao, W.; Wei, L.-S.; Li, Y.-Q.; Wang, W.; Wang, Y.; Zhou, Y.-W.; Zheng, Y.-Y.; et al. Distinct Roles of Smooth Muscle and Non-muscle Myosin Light Chain-Mediated Smooth Muscle Contraction. Front. Physiol. 2020, 11, 593966. [Google Scholar] [CrossRef]
- Hadadi, E.; Zhang, B.; Baidžajevas, K.; Yusof, N.; Puan, K.J.; Ong, S.M.; Yeap, W.H.; Rotzschke, O.; Kiss-Toth, E.; Wilson, H.; et al. Differential IL-1beta secretion by monocyte subsets is regulated by Hsp27 through modulating mRNA stability. Sci. Rep. 2016, 6, 39035. [Google Scholar] [CrossRef]
- Etzerodt, A.; Moestrup, S.K. CD163 and inflammation: Biological, diagnostic, and therapeutic aspects. Antioxid. Redox Signal. 2013, 18, 2352–2363. [Google Scholar] [CrossRef] [PubMed]
- Nikovics, K.; Favier, A.L. Macrophage Identification In Situ. Biomedicines 2021, 9, 1393. [Google Scholar] [CrossRef]
- Jiang, Y.-Q.; Wang, Z.-X.; Zhong, M.; Shen, L.-J.; Han, X.; Zou, X.; Liu, X.-Y.; Deng, Y.-N.; Yang, Y.; Che, G.-H.; et al. Investigating Mechanisms of Response or Resistance to Immune Checkpoint Inhibitors by Analyzing Cell-Cell Communications in Tumors Before and After Programmed Cell Death-1 (PD-1) Targeted Therapy: An Integrative Analysis Using Single-cell RNA and Bulk-RNA Sequencing Data. Oncoimmunology 2021, 10, 1908010. [Google Scholar] [PubMed]
- Saalbach, A.; Tremel, J.; Herbert, D.; Schwede, K.; Wandel, E.; Schirmer, C.; Anderegg, U.; Beck-Sickinger, A.G.; Heiker, J.T.; Schultz, S.; et al. Anti-Inflammatory Action of Keratinocyte-Derived Vaspin: Relevance for the Pathogenesis of Psoriasis. Am. J. Pathol. 2016, 186, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Sonawane, N.; Verkman, A.S. Localization of aquaporin-5 in sweat glands and functional analysis using knockout mice. J. Physiol. 2002, 541 Pt 2, 561–568. [Google Scholar] [CrossRef]
- Li, C.; Wang, S.; Chen, Y.; Zhang, X. Somatosensory Neuron Typing with High-Coverage Single-Cell RNA Sequencing and Functional Analysis. Neurosci. Bull. 2018, 34, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, A.; Kupcova, K.; Havranek, O. B-Cell Receptor Signaling and Beyond: The Role of Igalpha (CD79a)/Igbeta (CD79b) in Normal and Malignant B Cells. Int. J. Mol. Sci. 2023, 25, 10. [Google Scholar] [CrossRef]
- Theocharidis, G.; Baltzis, D.; Roustit, M.; Tellechea, A.; Dangwal, S.; Khetani, R.S.; Shu, B.; Zhao, W.; Fu, J.; Bhasin, S.; et al. Integrated Skin Transcriptomics and Serum Multiplex Assays Reveal Novel Mechanisms of Wound Healing in Diabetic Foot Ulcers. Diabetes 2020, 69, 2157–2169. [Google Scholar] [CrossRef]
- He, Y.; Tacconi, C.; Dieterich, L.C.; Kim, J.; Restivo, G.; Gousopoulos, E.; Lindenblatt, N.; Levesque, M.P.; Claassen, M.; Detmar, M. Novel Blood Vascular Endothelial Subtype-Specific Markers in Human Skin Unearthed by Single-Cell Transcriptomic Profiling. Cells 2022, 11, 1111. [Google Scholar] [CrossRef]
- Huang, M.; Tabib, T.; Khanna, D.; Assassi, S.; Domsic, R.; Lafyatis, R. Single-cell transcriptomes and chromatin accessibility of endothelial cells unravel transcription factors associated with dysregulated angiogenesis in systemic sclerosis. Ann. Rheum. Dis. 2024. online ahead of print. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Z.; Wang, W.; Liu, M.; Bao, Y. Differential effects of sulforaphane in regulation of angiogenesis in a co-culture model of endothelial cells and pericytes. Oncol. Rep. 2017, 37, 2905–2912. [Google Scholar] [CrossRef]
- Jin, Y.; An, X.; Ye, Z.; Cully, B.; Wu, J.; Li, J. RGS5, a hypoxia-inducible apoptotic stimulator in endothelial cells. J. Biol. Chem. 2009, 284, 23436–23443. [Google Scholar] [CrossRef]
- Quillard, T.; Charreau, B. Impact of notch signaling on inflammatory responses in cardiovascular disorders. Int. J. Mol. Sci. 2013, 14, 6863–6888. [Google Scholar] [CrossRef] [PubMed]
- Zahoor, M.A.; Xue, G.; Sato, H.; Murakami, T.; Takeshima, S.-N.; Aida, Y. HIV-1 Vpr induces interferon-stimulated genes in human monocyte-derived macrophages. PLoS ONE 2014, 9, e106418. [Google Scholar] [CrossRef] [PubMed]
- Gratton, R.; Tricarico, P.M.; Pio d, A.; Bianco, A.M.; Moura, R.; Agrelli, A.; Brandão, L.; Zupin, L.; Crovella, S. Notch Signaling Regulation in Autoinflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 8847. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenberg, J.F.; Smeets, E.F.; Neefjes, J.J.; Shaffer, M.A.; Cinek, T.; Jeunhomme, T.M.; Ahern, T.J.; Buurman, W.A. E-selectin and intercellular adhesion molecule-1 are released by activated human endothelial cells in vitro. Immunology 1992, 77, 543–549. [Google Scholar] [PubMed]
- Milstone, D.S.; Fukumura, D.; Padgett, R.C.; O’Donnell, P.E.; Davis, V.M.; Benavidez, O.J.; Monsky, W.L.; Melder, R.J.; Jain, R.K.; Gimbrone, M.A., Jr. Mice lacking E-selectin show normal numbers of rolling leukocytes but reduced leukocyte stable arrest on cytokine-activated microvascular endothelium. Microcirculation 1998, 5, 153–171. [Google Scholar]
- Ramos, C.L.; Kunkel, E.J.; Lawrence, M.B.; Jung, U.; Vestweber, D.; Bosse, R.; McIntyre, K.W.; Gillooly, K.M.; Norton, C.R.; Wolitzky, B.A.; et al. Differential effect of E-selectin antibodies on neutrophil rolling and recruitment to inflammatory sites. Blood 1997, 89, 3009–3018. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, E.J.; Dunne, J.L.; Ley, K. Leukocyte arrest during cytokine-dependent inflammation in vivo. J. Immunol. 2000, 164, 3301–3308. [Google Scholar] [CrossRef]
- Ley, K.; Allietta, M.; Bullard, D.C.; Morgan, S. Importance of E-selectin for firm leukocyte adhesion in vivo. Circ Res. 1998, 83, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Jin, Y.; Lin, M.; Xia, X.; Chen, X.; Huang, A. Down-regulation of Xist and Mir-7a-5p improves LPS-induced myocardial injury. Int. J. Med. Sci. 2020, 17, 2570–2577. [Google Scholar] [CrossRef]
- Dou, D.R.; Zhao, Y.; Belk, J.A.; Zhao, Y.; Casey, K.M.; Chen, D.C.; Li, R.; Yu, B.; Srinivasan, S.; Abe, B.T.; et al. Xist ribonucleoproteins promote female sex-biased autoimmunity. Cell 2024, 187, 733–749.e16. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J. Interleukin-33 (IL-33): A critical review of its biology and the mechanisms involved in its release as a potent extracellular cytokine. Cytokine 2022, 156, 155891. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Oka, T.; Demehri, S. Alarmin Cytokines as Central Regulators of Cutaneous Immunity. Front. Immunol. 2022, 13, 876515. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1alpha, IL-33 and S100 proteins: Dual-function alarmins. Cell Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef]
- Pendergrass, S.A.; Hayes, E.; Farina, G.; Lemaire, R.; Farber, H.W.; Whitfield, M.L.; Lafyatis, R. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS ONE 2010, 5, e12106. [Google Scholar] [CrossRef] [PubMed]
- Mecoli, C.A.; Shah, A.A.; Boin, F.; Wigley, F.M.; Hummers, L.K. The Utility of Plasma Vascular Biomarkers in Systemic Sclerosis: A Prospective Longitudinal Analysis. Arthritis Rheumatol. 2020, 72, 1341–1349. [Google Scholar] [CrossRef]
- Kawashiri, S.-Y.; Nishino, A.; Igawa, T.; Takatani, A.; Shimizu, T.; Umeda, M.; Fukui, S.; Okada, A.; Suzuki, T.; Koga, T.; et al. Prediction of organ involvement in systemic sclerosis by serum biomarkers and peripheral endothelial function. Clin. Exp. Rheumatol. 2018, 36 (Suppl. S113), 102–108. [Google Scholar] [PubMed]
- Gur, C.; Wang, S.-Y.; Sheban, F.; Zada, M.; Li, B.; Kharouf, F.; Peleg, H.; Aamar, S.; Yalin, A.; Kirschenbaum, D.; et al. LGR5 expressing skin fibroblasts define a major cellular hub perturbed in scleroderma. Cell 2022, 185, 1373–1388.e20. [Google Scholar] [CrossRef] [PubMed]
- Mirizio, E.; Marathi, A.; Hershey, N.; Ross, C.; Schollaert, K.; Salgado, C.; Reyes-Mugica, M.; Torok, K.S. Identifying the Signature Immune Phenotypes Present in Pediatric Localized Scleroderma. J. Investig. Dermatol. 2019, 139, 715–718. [Google Scholar] [CrossRef]
- O’Brien, J.C.; Rainwater, Y.B.; Malviya, N.; Cyrus, N.; Auer-Hackenberg, L.; Hynan, L.S.; Hosler, G.A.; Jacobe, H.T. Transcriptional and Cytokine Profiles Identify CXCL9 as a Biomarker of Disease Activity in Morphea. J. Investig. Dermatol. 2017, 137, 1663–1670. [Google Scholar]
- Spasovski, V.; Andjelkovic, M.; Parezanovic, M.; Komazec, J.; Ugrin, M.; Klaassen, K.; Stojiljkovic, M. The Role of Autophagy and Apoptosis in Affected Skin and Lungs in Patients with Systemic Sclerosis. Int. J. Mol. Sci. 2023, 24, 11212. [Google Scholar] [CrossRef]
- Lai, E.C. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef]
- Garrison, G.; Huang, S.K.; Okunishi, K.; Scott, J.P.; Penke, L.R.K.; Scruggs, A.M.; Peters-Golden, M. Reversal of myofibroblast differentiation by prostaglandin E(2). Am. J. Respir. Cell Mol. Biol. 2013, 48, 550–558. [Google Scholar] [CrossRef]
- Dees, C.; Tomcik, M.; Zerr, P.; Akhmetshina, A.; Horn, A.; Palumbo, K.; Beyer, C.; Zwerina, J.; Distler, O.; Schett, G.; et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 1304–1310. [Google Scholar] [CrossRef]
- Choi, A.; Nam, S.A.; Kim, W.-Y.; Park, S.H.; Kim, H.; Yang, C.W.; Kim, J.; Kim, Y.K. Notch signaling in the collecting duct regulates renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction in mice. Korean J. Intern. Med. 2018, 33, 774–782. [Google Scholar] [CrossRef]
- Ciechomska, M.; van Laar, J.; O’Reilly, S. Current frontiers in systemic sclerosis pathogenesis. Exp. Dermatol. 2015, 24, 401–406. [Google Scholar] [CrossRef]
- Amsen, D.; Blander, J.M.; Lee, G.R.; Tanigaki, K.; Honjo, T.; Flavell, R.A. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell 2004, 117, 515–526. [Google Scholar] [CrossRef]
- Bielesz, B.; Sirin, Y.; Si, H.; Niranjan, T.; Gruenwald, A.; Ahn, S.; Kato, H.; Pullman, J.; Gessler, M.; Haase, V.H. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J. Clin. Investig. 2010, 120, 4040–4054. [Google Scholar] [CrossRef]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.C.; et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat. Commun. 2020, 11, 2042. [Google Scholar] [CrossRef]
- Kawano-Dourado, L.; Ab, A.M.; Capelozzi, V.L.; Valeri, C.; Barbas, C.S.V. In situ evidence of pulmonary endothelial activation in patients with granulomatosis with polyangiitis and systemic sclerosis. Lung 2015, 193, 355–359. [Google Scholar] [CrossRef]
- Mouawad, J.E.; Feghali-Bostwick, C. The Molecular Mechanisms of Systemic Sclerosis-Associated Lung Fibrosis. Int. J. Mol. Sci. 2023, 24, 2963. [Google Scholar] [CrossRef]
- Yanaba, K.; Yoshizaki, A.; Asano, Y.; Kadono, T.; Sato, S. Serum IL-33 levels are raised in patients with systemic sclerosis: Association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin. Rheumatol. 2011, 30, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Grönberg, C.; Rattik, S.; Tran-Manh, C.; Zhou, X.; Rigau, A.R.; Li, Y.-N.; Györfi, A.-H.; Dickel, N.; Kunz, M.; Kreuter, A.; et al. Combined inhibition of IL-1, IL-33 and IL-36 signalling by targeting IL1RAP ameliorates skin and lung fibrosis in preclinical models of systemic sclerosis. Ann. Rheum. Dis. 2024, 2024. 83, 1156–1168. [Google Scholar] [CrossRef]
- England, E.; Rees, D.G.; Scott, I.C.; Carmen, S.; Chan, D.T.Y.; Huntington, C.E.C.; Houslay, K.F.; Erngren, T.; Penney, M.; Majithiy, J.B.; et al. Tozorakimab (MEDI3506): An anti-IL-33 antibody that inhibits IL-33 signalling via ST2 and RAGE/EGFR to reduce inflammation and epithelial dysfunction. Sci. Rep. 2023, 13, 9825. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Wagner, B.; Zucchetto, M.; Raghu, G.; Martinez, F.J.; Goldin, J.; Siegel, J.; Denton, C.P. Long-Term Safety and Efficacy of Tocilizumab in Early Systemic Sclerosis-Interstitial Lung Disease: Open-Label Extension of a Phase 3 Randomized Controlled Trial. Am. J. Respir. Crit Care Med. 2022, 205, 674–684. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, J.; Yin, Y.; Li, K.; Zhang, C.; Zheng, Y. The Role of IL-6 in Fibrotic Diseases: Molecular and Cellular Mechanisms. Int. J. Biol. Sci. 2022, 18, 5405–5414. [Google Scholar] [CrossRef] [PubMed]
- Shingu, M.; Miyauchi, S.; Nagai, Y.; Yasutake, C.; Horie, K. The role of IL-4 and IL-6 in IL-1-dependent cartilage matrix degradation. Br. J. Rheumatol. 1995, 34, 101–106. [Google Scholar] [CrossRef]
- Rose-John, S.; Jenkins, B.J.; Garbers, C.; Moll, J.M.; Scheller, J. Targeting IL-6 trans-signalling: Past, present and future prospects. Nat. Rev. Immunol. 2023, 23, 666–681. [Google Scholar] [CrossRef]
- O’Reilly, S.; Cant, R.; Ciechomska, M.; van Laar, J.M. Interleukin-6: A new therapeutic target in systemic sclerosis? Clin. Transl. Immunol. 2013, 2, e4. [Google Scholar] [CrossRef]
- Jin, J.; Liu, Y.; Tang, Q.; Yan, X.; Jiang, M.; Zhao, X.; Chen, J.; Jin, C.; Ou, Q.; Zhao, J. Bioinformatics-integrated screening of systemic sclerosis-specific expressed markers to identify therapeutic targets. Front. Immunol. 2023, 14, 1125183. [Google Scholar] [CrossRef]
- Almalki, W.H. Unraveling the role of Xist RNA in cardiovascular pathogenesis. Pathol. Res. Pract. 2024, 253, 154944. [Google Scholar] [CrossRef]
- Hu, C.; Bai, X.; Liu, C.; Hu, Z. Long noncoding RNA XIST participates hypoxia-induced angiogenesis in human brain microvascular endothelial cells through regulating miR-485/SOX7 axis. Am. J. Transl. Res. 2019, 11, 6487–6497. [Google Scholar] [PubMed]
- Wang, C.; Dong, J.; Sun, J.; Huang, S.; Wu, F.; Zhang, X.; Pang, D.; Fu, Y.; Li, L. Silencing of lncRNA XIST impairs angiogenesis and exacerbates cerebral vascular injury after ischemic stroke. Mol. Ther. Nucleic Acids 2021, 26, 148–160. [Google Scholar] [CrossRef]
- Schäbitz, A.; Hillig, C.; Mubarak, M.; Jargosch, M.; Farnoud, A.; Scala, E.; Kurzen, N.; Pilz, A.C.; Bhalla, N.; Thomas, J.; et al. Spatial transcriptomics landscape of lesions from non-communicable inflammatory skin diseases. Nat. Commun. 2022, 13, 7729. [Google Scholar] [CrossRef]
- Squair, J.W.; Gautier, M.; Kathe, C.; Anderson, M.A.; James, N.D.; Hutson, T.H.; Hudelle, R.; Qaiser, T.; Matson, K.J.E.; Barraud, Q.; et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021, 12, 5692. [Google Scholar] [CrossRef]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutchins, T.; Sanyal, A.; Esencan, D.; Lafyatis, R.; Jacobe, H.; Torok, K.S. Characterization of Endothelial Cell Subclusters in Localized Scleroderma Skin with Single-Cell RNA Sequencing Identifies NOTCH Signaling Pathway. Int. J. Mol. Sci. 2024, 25, 10473. https://doi.org/10.3390/ijms251910473
Hutchins T, Sanyal A, Esencan D, Lafyatis R, Jacobe H, Torok KS. Characterization of Endothelial Cell Subclusters in Localized Scleroderma Skin with Single-Cell RNA Sequencing Identifies NOTCH Signaling Pathway. International Journal of Molecular Sciences. 2024; 25(19):10473. https://doi.org/10.3390/ijms251910473
Chicago/Turabian StyleHutchins, Theresa, Anwesha Sanyal, Deren Esencan, Robert Lafyatis, Heidi Jacobe, and Kathryn S. Torok. 2024. "Characterization of Endothelial Cell Subclusters in Localized Scleroderma Skin with Single-Cell RNA Sequencing Identifies NOTCH Signaling Pathway" International Journal of Molecular Sciences 25, no. 19: 10473. https://doi.org/10.3390/ijms251910473