
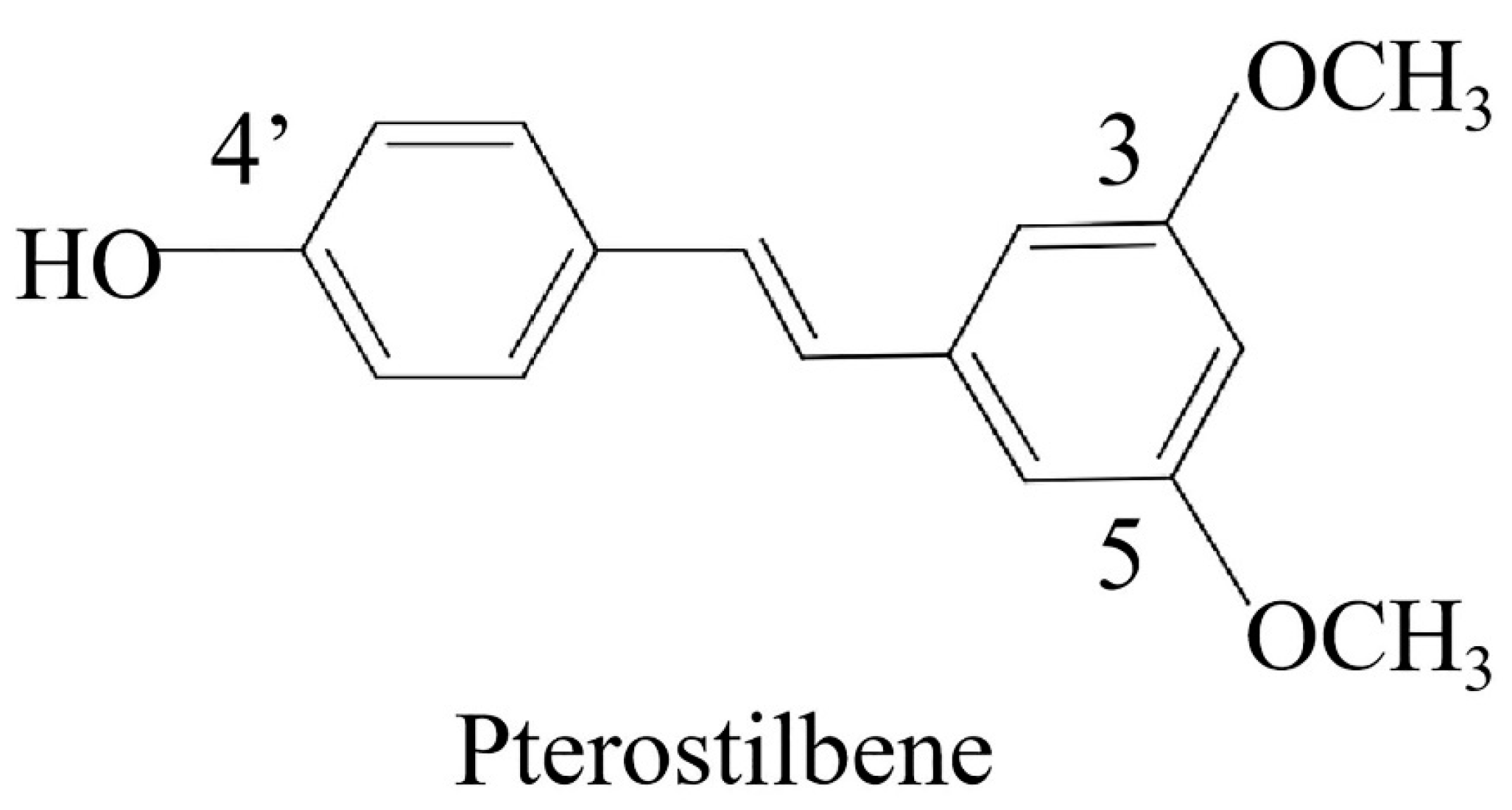
Pterostilbene Induces Pyroptosis in Breast Cancer Cells through Pyruvate Kinase 2/Caspase-8/Gasdermin C Signaling Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
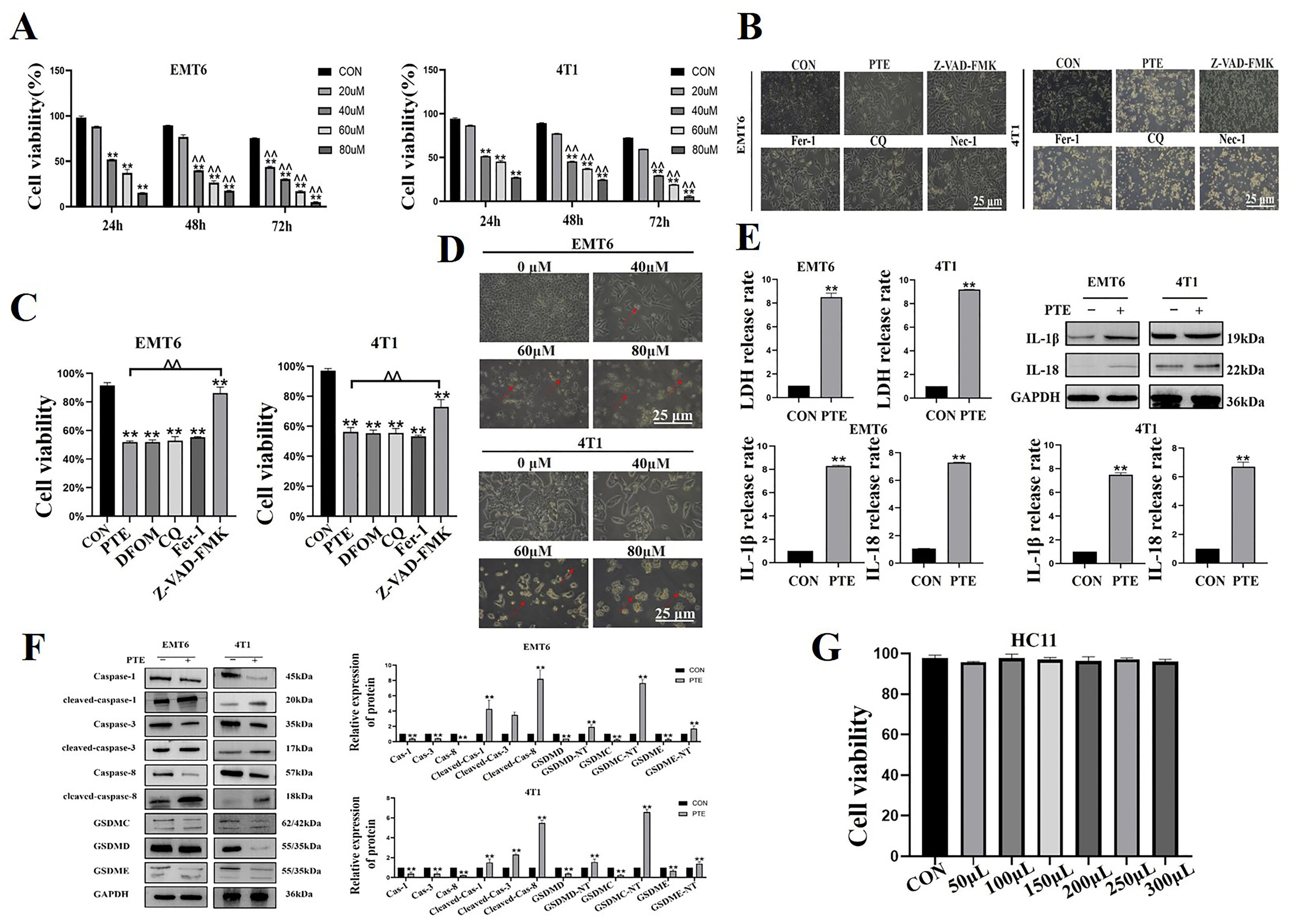
2.1. PTE-Induced Pyroptosis in EMT6 and 4T1 Cells
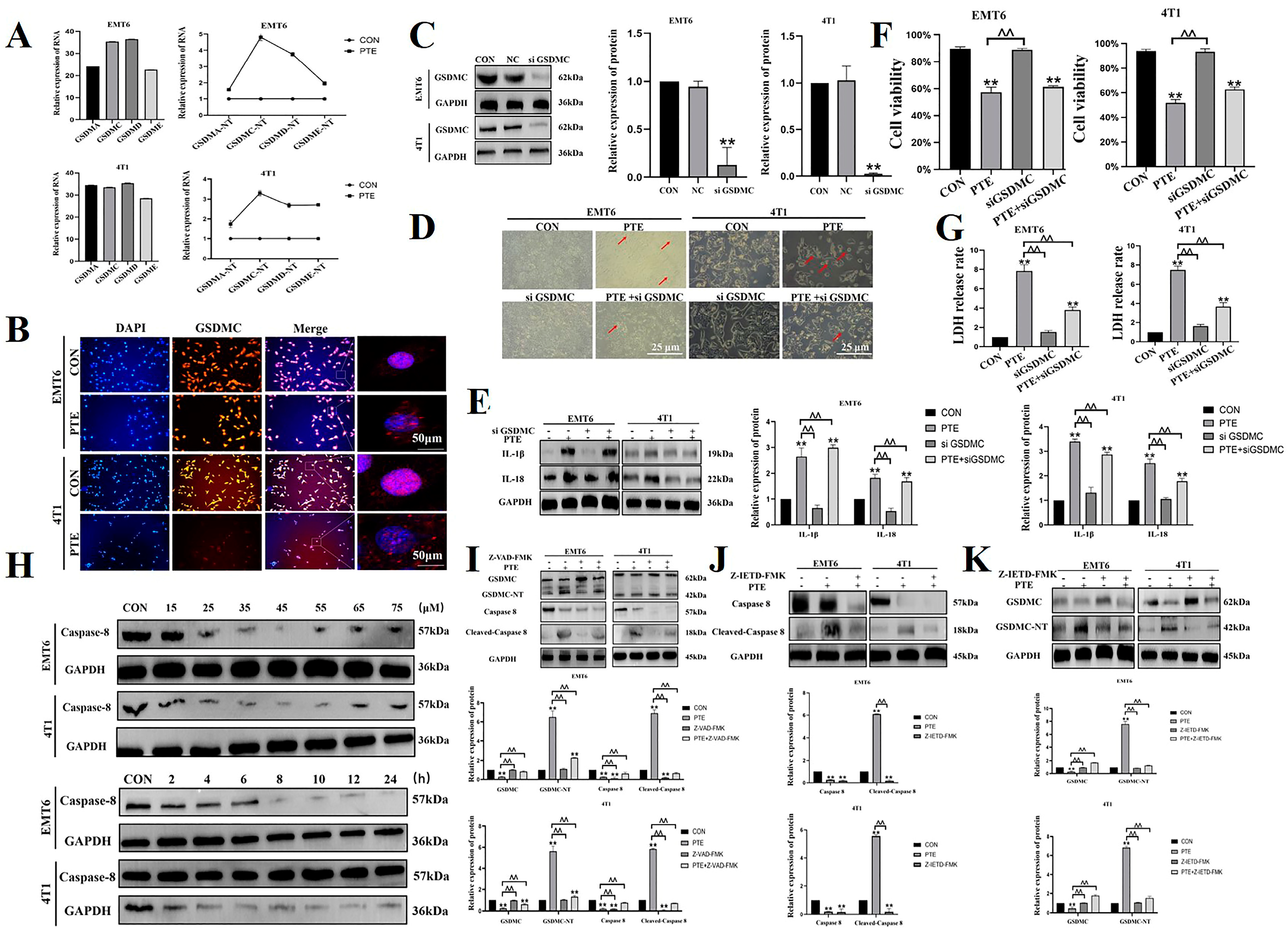
2.2. PTE-Induced Cancer Cell Pyroptosis Is Mediated by GSDMC
2.3. PTE Inhibits the Glycolysis of EMT6 and 4T1 Cells
2.4. PTE Inhibits Glycolysis by Regulating PKM2, a Key Rate-Limiting Enzyme in Glycolysis
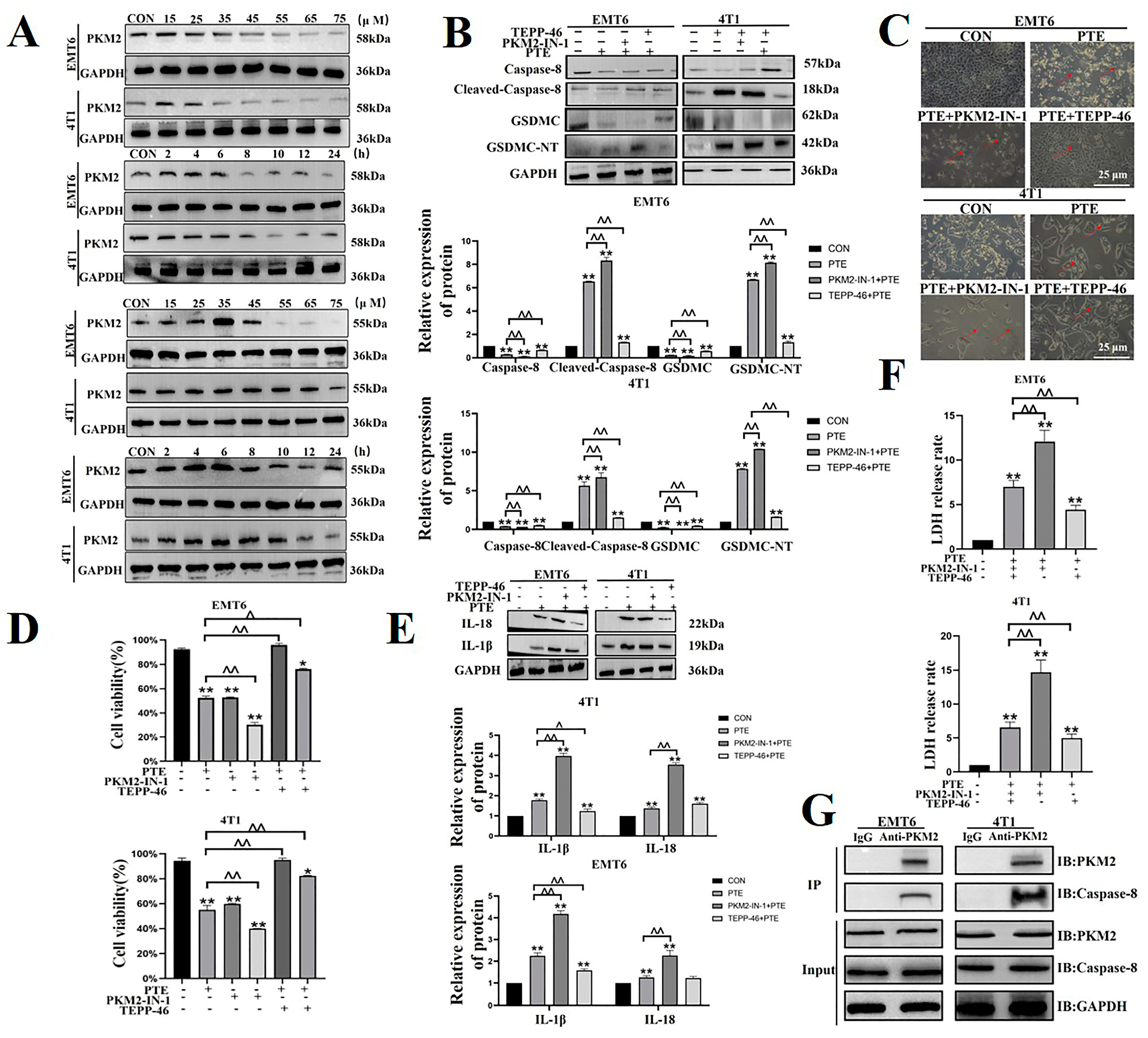
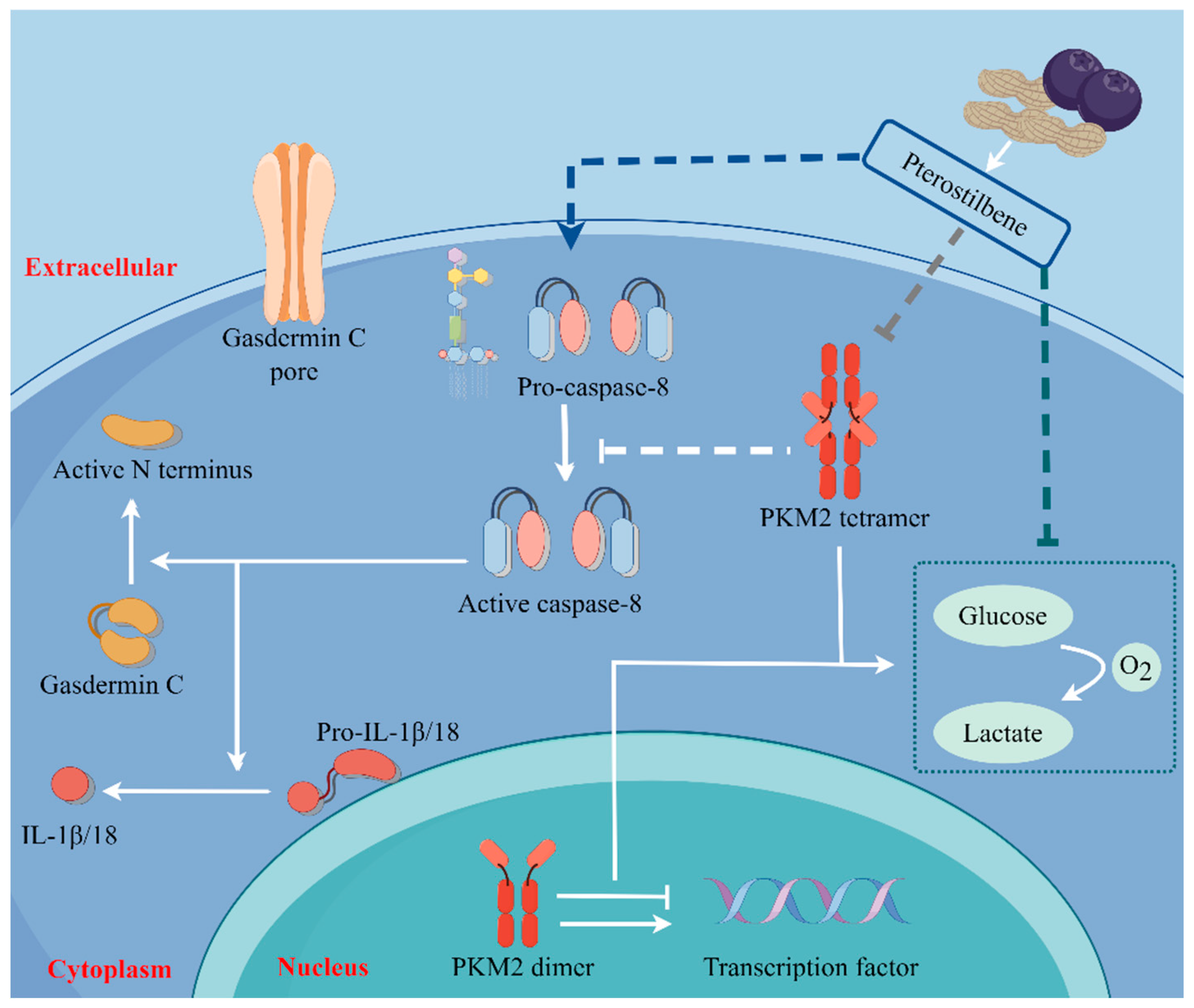
2.5. PTE-Activated Caspase-8/GSDMC Cascade through Repressing PKM2
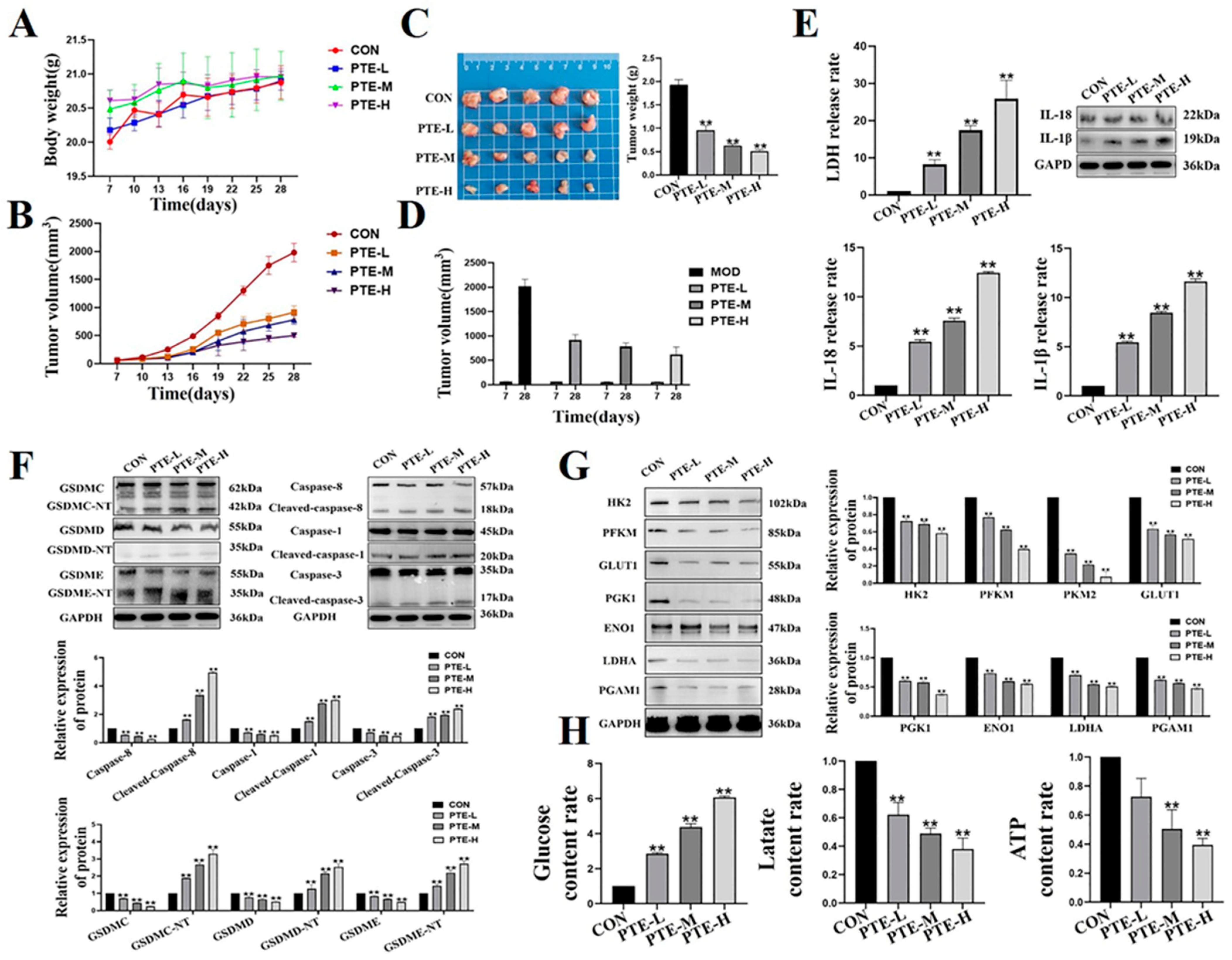
2.6. PTE Reduces Tumorigenicity in Mouse Breast Tumor Models
3. Discussion
4. Methods
4.1. Cell Culture
4.2. Test Animals
4.3. Preparation Solutions
4.4. Cell Counting Kit-8 (CCK8) Assays
4.5. Morphological Observations
4.6. Real-Time Fluorescence Quantitative PCR (qPCR)
4.7. Western Blot (WB)
4.8. Immunofluorescence
4.9. DSS Cross-Linking Test
4.10. Transfection
4.11. Nucleoplasmic Separation
4.12. Pyruvate Kinase Activity
4.13. Glucose, Lactate, and Adenosine Triphosphate (ATP) Detection
4.14. Co-IP
4.15. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| adenosine triphosphate | ATP |
| Cell Counting Kit-8 | CCK8 |
| Dimethyl sulfoxide | DMSO |
| Gasdermins | GSDMs |
| hexokinase 2 | HK2 |
| interleukin | IL |
| Lactate dehydrogenase | LDH |
| Pterostilbene | PTE |
| phosphofructokinase-1 | PFK-1 |
| phosphate-buffered saline | PBS, pH 8.0 |
| pyruvate kinase | PK |
| standard deviation | SD |
| Western Blot | WB |
References
- Fan, L.; Strasser-Weippl, K.; Li, J.J.; Louis, J.S.; Finkelstein, D.M.; Yu, K.D.; Chen, W.Q.; Shao, Z.M.; Goss, P.E. Breast cancer in China. Lancet Oncol. 2014, 15, e279–e289. [Google Scholar] [CrossRef] [PubMed]
- Moo, T.A.; Sanford, R.; Dang, C.; Morrow, M. Overview of Breast Cancer Therapy. PET Clin. 2018, 13, 339–354. [Google Scholar] [CrossRef]
- Anampa, J.; Makower, D.; Sparano, J.A. Progress in adjuvant chemotherapy for breast cancer: An overview. BMC Med. 2015, 13, 195. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 2021, 12, 1786. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, X.; Zhang, Y. Biological Functions of Gasdermins in Cancer: From Molecular Mechanisms to Therapeutic Potential. Front. Cell Dev. Biol. 2021, 9, 638710. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Fox, D.; Man, S.M. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J. Mol. Biol. 2018, 430, 3068–3080. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 5, 28. [Google Scholar] [CrossRef]
- Cardoso, M.R.; Santos, J.C.; Ribeiro, M.L.; Talarico, M.C.R.; Viana, L.R.; Derchain, S.F.M. A Metabolomic Approach to Predict Breast Cancer Behavior and Chemotherapy Response. Int. J. Mol. Sci. 2018, 19, 617. [Google Scholar] [CrossRef]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Verma, H.; Cholia, R.P.; Kaur, S.; Dhiman, M.; Mantha, A.K. A short review on cross-link between pyruvate kinase (PKM2) and Glioblastoma Multiforme. Metab. Brain Dis. 2021, 36, 751–765. [Google Scholar] [CrossRef]
- Puckett, D.L.; Alquraishi, M.; Chowanadisai, W.; Bettaieb, A. The Role of PKM2 in Metabolic Reprogramming: Insights into the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 1171. [Google Scholar] [CrossRef] [PubMed]
- Duta-Bratu, C.G.; Nitulescu, G.M.; Mihai, D.P.; Olaru, O.T. Resveratrol and Other Natural Oligomeric Stilbenoid Compounds and Their Therapeutic Applications. Plants 2023, 12, 2935. [Google Scholar] [CrossRef] [PubMed]
- Estrela, J.M.; Ortega, A.; Mena, S.; Rodriguez, M.L.; Asensi, M. Pterostilbene: Biomedical applications. Crit. Rev. Clin. Lab. Sci. 2013, 50, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Xiao, W.; Gu, W.T.; Zhang, Z.T.; Xu, S.H.; Chen, Z.Q.; Xu, Y.H.; Zhang, L.Y.; Wang, S.M.; Nie, H. Pterostilbene prevents methylglyoxal-induced cytotoxicity in endothelial cells by regulating glyoxalase, oxidative stress and apoptosis. Food. Chem. Toxicol. 2021, 153, 112244. [Google Scholar] [CrossRef]
- Li, Q.; Chen, L.; Liu, X.; Li, X.; Cao, Y.; Bai, Y.; Qi, F. Pterostilbene inhibits amyloid-β-induced neuroinflammation in a microglia cell line by inactivating the NLRP3/caspase-1 inflammasome pathway. J. Cell. Biochem. 2018, 119, 7053–7062. [Google Scholar] [CrossRef]
- Gao, H.; Liu, Z.; Xu, W.; Wang, Q.; Zhang, C.; Ding, Y.; Nie, W.; Lai, J.; Chen, Y.; Huang, H. Pterostilbene promotes mitochondrial apoptosis and inhibits proliferation in glioma cells. Sci. Rep. 2021, 11, 6381. [Google Scholar] [CrossRef]
- Chang, H.P.; Lu, C.C.; Chiang, J.H.; Tsai, F.J.; Juan, Y.N.; Tsao, J.W.; Chiu, H.Y.; Yang, J.S. Pterostilbene modulates the suppression of multidrug resistance protein 1 and triggers autophagic and apoptotic mechanisms in cisplatin-resistant human oral cancer CAR cells via AKT signaling. Int. J. Oncol. 2018, 52, 1504–1514. [Google Scholar]
- Zhang, J.Y.; Zhou, B.; Sun, R.Y.; Ai, Y.L.; Cheng, K.; Li, F.N.; Wang, B.R.; Liu, F.J.; Jiang, Z.H.; Wang, W.J.; et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 2021, 31, 980–997. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Sato, T.; Nomura, M.; Sakamoto, Y.; Inoue, Y.; Tanaka, R.; Ito, S.; Kurosawa, K.; Yamaguchi, K.; Sugiura, Y.; et al. PKM1 Confers Metabolic Advantages and Promotes Cell-Autonomous Tumor Cell Growth. Cancer Cell 2018, 33, 355–367.e357. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, P.; Li, Z. The multifaceted regulation and functions of PKM2 in tumor progression. Biochim. Et Biophys. Acta 2014, 1846, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Radecka, B.; Litwiniuk, M. Breast cancer in young women. Ginekol. Pol. 2016, 87, 659–663. [Google Scholar] [CrossRef]
- Tan, J.; Le, A. Breast Cancer Metabolism. Adv. Exp. Med. Biol. 2018, 1063, 83–93. [Google Scholar]
- Liao, M.; Qin, R.; Huang, W.; Zhu, H.P.; Peng, F.; Han, B.; Liu, B. Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: A revisited perspective from molecular mechanisms to targeted therapies. J. Hematol. Oncol. 2022, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wang, Z.; Dai, P.; Sun, J.; Li, J.; Han, W.; Li, K. The activation of SIRT1 by resveratrol reduces breast cancer metastasis to lung through inhibiting neutrophil extracellular traps. J. Drug Target. 2023, 31, 962–975. [Google Scholar] [CrossRef]
- Yang, H.; Zheng, Y.; Li, T.W.; Peng, H.; Fernandez-Ramos, D.; Martínez-Chantar, M.L.; Rojas, A.L.; Mato, J.M.; Lu, S.C. Methionine adenosyltransferase 2B, HuR, and sirtuin 1 protein cross-talk impacts on the effect of resveratrol on apoptosis and growth in liver cancer cells. J. Biol. Chem. 2013, 288, 23161–23170. [Google Scholar] [CrossRef]
- Li, N.; Sun, C.; Zhou, B.; Xing, H.; Ma, D.; Chen, G.; Weng, D. Low concentration of quercetin antagonizes the cytotoxic effects of anti-neoplastic drugs in ovarian cancer. PLoS ONE 2014, 9, e100314. [Google Scholar] [CrossRef]
- Zhang, X.A.; Zhang, S.; Yin, Q.; Zhang, J. Quercetin induces human colon cancer cells apoptosis by inhibiting the nuclear factor-kappa B Pathway. Pharmacogn. Mag. 2015, 11, 404–409. [Google Scholar] [CrossRef]
- Wang, P.; Sang, S. Metabolism and pharmacokinetics of resveratrol and pterostilbene. BioFactors 2018, 44, 16–25. [Google Scholar] [CrossRef]
- Oruganti, L.; Meriga, B. Plant Polyphenolic Compounds Potentiates Therapeutic Efficiency of Anticancer Chemotherapeutic Drugs: A Review. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.J.; Fernández, M.; Picó, Y.; Mañes, J.; Asensi, M.; Carda, C.; Asensio, G.; Estrela, J.M. Dietary administration of high doses of pterostilbene and quercetin to mice is not toxic. J. Agric. Food Chem. 2009, 57, 3180–3186. [Google Scholar] [CrossRef]
- Lin, Y.K.; Yeh, C.T.; Kuo, K.T.; Yadav, V.K.; Fong, I.H.; Kounis, N.G.; Hu, P.; Hung, M.Y. Pterostilbene Increases LDL Metabolism in HL-1 Cardiomyocytes by Modulating the PCSK9/HNF1α/SREBP2/LDLR Signaling Cascade, Upregulating Epigenetic hsa-miR-335 and hsa-miR-6825, and LDL Receptor Expression. Antioxidants 2021, 10, 1280. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Y.; Chen, Y.; Ji, S.; Jia, P.; Xu, J.; Li, Y.; Wang, T. Pterostilbene attenuates liver injury and oxidative stress in intrauterine growth-retarded weanling piglets. Nutrition 2021, 81, 110940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Han, Z.; Jiang, A.; Wu, D.; Li, S.; Liu, Z.; Wei, Z.; Yang, Z.; Guo, C. Protective Effects of Pterostilbene on Lipopolysaccharide-Induced Acute Lung Injury in Mice by Inhibiting NF-κB and Activating Nrf2/HO-1 Signaling Pathways. Front. Pharmacol. 2020, 11, 591836. [Google Scholar] [CrossRef] [PubMed]
- Pimentel-Moral, S.; Teixeira, M.C.; Fernandes, A.R.; Arráez-Román, D.; Martínez-Férez, A.; Segura-Carretero, A.; Souto, E.B. Lipid nanocarriers for the loading of polyphenols—A comprehensive review. Adv. Colloid Interface Sci. 2018, 260, 85–94. [Google Scholar] [CrossRef]
- Pan, C.; Hu, Y.; Li, J.; Wang, Z.; Huang, J.; Zhang, S.; Ding, L. Estrogen receptor-α36 is involved in pterostilbene-induced apoptosis and anti-proliferation in in vitro and in vivo breast cancer. PLoS ONE 2014, 9, e104459. [Google Scholar] [CrossRef]
- Kala, R.; Tollefsbol, T.O. A Novel Combinatorial Epigenetic Therapy Using Resveratrol and Pterostilbene for Restoring Estrogen Receptor-α (ERα) Expression in ERα-Negative Breast Cancer Cells. PLoS ONE 2016, 11, e0155057. [Google Scholar] [CrossRef]
- Surien, O.; Ghazali, A.R.; Masre, S.F. Chemopreventive effects of pterostilbene through p53 and cell cycle in mouse lung of squamous cell carcinoma model. Sci. Rep. 2021, 11, 14862. [Google Scholar] [CrossRef]
- Priego, S.; Feddi, F.; Ferrer, P.; Mena, S.; Benlloch, M.; Ortega, A.; Carretero, J.; Obrador, E.; Asensi, M.; Estrela, J.M. Natural polyphenols facilitate elimination of HT-29 colorectal cancer xenografts by chemoradiotherapy: A Bcl-2- and superoxide dismutase 2-dependent mechanism. Mol. Cancer Ther. 2008, 7, 3330–3342. [Google Scholar] [CrossRef]
- Locquet, M.A.; Ichim, G.; Bisaccia, J.; Dutour, A.; Lebecque, S.; Castets, M.; Weber, K. Caspase-8 deficiency induces a switch from TLR3 induced apoptosis to lysosomal cell death in neuroblastoma. Sci. Rep. 2021, 11, 10609. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Deng, W.; Bai, Y.; Miao, R.; Mei, S.; Zhang, Z.; Pan, Y.; Wang, Y.; Min, R.; Deng, F.; et al. The Lysosomal Rag-Ragulator Complex Licenses RIPK1 and Caspase-8-mediated Pyroptosis by Yersinia. Science 2021, 372, eabg0269. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, X.; Huang, S.; Chen, J.; Ding, P.; Wang, Q.; Li, L.; Lv, X.; Li, L.; Zhang, P.; et al. FOXM1D potentiates PKM2-mediated tumor glycolysis and angiogenesis. Mol. Oncol. 2021, 15, 1466–1485. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Zhu, W.; Han, J.; Yang, X.; Zhou, R.; Lu, H.C.; Yu, H.; Yuan, W.B.; Li, P.C.; Tao, J.; et al. The role of the HIF-1α/ALYREF/PKM2 axis in glycolysis and tumorigenesis of bladder cancer. Cancer Commun. 2021, 41, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tong, L.; Luo, Y.; Li, X.; Chen, G.; Wang, Y. Resveratrol inhibits the proliferation and induces the apoptosis in ovarian cancer cells via inhibiting glycolysis and targeting AMPK/mTOR signaling pathway. J. Cell. Biochem. 2018, 119, 6162–6172. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, A.; Zhang, L.; Zhang, J.; Zhong, Z.; Wang, F. Knockdown of PKM2 Suppresses Tumor Growth and Invasion in Lung Adenocarcinoma. Int. J. Mol. Sci. 2015, 16, 24574–24587. [Google Scholar] [CrossRef]
- Guan, M.; Tong, Y.; Guan, M.; Liu, X.; Wang, M.; Niu, R.; Zhang, F.; Dong, D.; Shao, J.; Zhou, Y. Lapatinib Inhibits Breast Cancer Cell Proliferation by Influencing PKM2 Expression. Technol. Cancer Res. Treat. 2018, 17, 1533034617749418. [Google Scholar] [CrossRef]
- Shan, S.; Shi, J.; Yang, P.; Jia, B.; Wu, H.; Zhang, X.; Li, Z. Apigenin Restrains Colon Cancer Cell Proliferation via Targeted Blocking of Pyruvate Kinase M2-Dependent Glycolysis. J. Agric. Food Chem. 2017, 65, 8136–8144. [Google Scholar] [CrossRef]
- Sanman, L.E.; Qian, Y.; Eisele, N.A.; Ng, T.M.; van der Linden, W.A.; Monack, D.M.; Weerapana, E.; Bogyo, M. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. eLife 2016, 5, e13663. [Google Scholar] [CrossRef]
- Deng, R.; Wang, S.M.; Yin, T.; Ye, T.H.; Shen, G.B.; Li, L.; Zhao, J.Y.; Sang, Y.X.; Duan, X.G.; Wei, Y.Q. Dimethyl Sulfoxide Suppresses Mouse 4T1 Breast Cancer Growth by Modulating Tumor-Associated Macrophage Differentiation. J. Breast Cancer 2014, 17, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, X.; Wang, P.; Zhang, K.; Wang, H.; Feng, X.; Liu, Q. Efficacy of chlorin e6-mediated sono-photodynamic therapy on 4T1 cells. Cancer Biother. Radiopharm. 2014, 29, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.E.; Vellanki, S.H.; Smith, Y.E.; Hopkins, A.M. Diterpenoid natural compound C4 (Crassin) exerts cytostatic effects on triple-negative breast cancer cells via a pathway involving reactive oxygen species. Cell. Oncol. 2018, 41, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, G.; Therriault, H.; Geha, S.; Bérubé-Lauzière, Y.; Bujold, R.; Saucier, C.; Paquette, B. Stimulation of triple negative breast cancer cell migration and metastases formation is prevented by chloroquine in a pre-irradiated mouse model. BMC Cancer 2016, 16, 361. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, T.; Peng, L.; Dong, J.; Li, L. Pterostilbene Induces Pyroptosis in Breast Cancer Cells through Pyruvate Kinase 2/Caspase-8/Gasdermin C Signaling Pathway. Int. J. Mol. Sci. 2024, 25, 10509. https://doi.org/10.3390/ijms251910509
Pan T, Peng L, Dong J, Li L. Pterostilbene Induces Pyroptosis in Breast Cancer Cells through Pyruvate Kinase 2/Caspase-8/Gasdermin C Signaling Pathway. International Journal of Molecular Sciences. 2024; 25(19):10509. https://doi.org/10.3390/ijms251910509
Chicago/Turabian StylePan, Tingting, Li Peng, Jing Dong, and Lin Li. 2024. "Pterostilbene Induces Pyroptosis in Breast Cancer Cells through Pyruvate Kinase 2/Caspase-8/Gasdermin C Signaling Pathway" International Journal of Molecular Sciences 25, no. 19: 10509. https://doi.org/10.3390/ijms251910509
APA StylePan, T., Peng, L., Dong, J., & Li, L. (2024). Pterostilbene Induces Pyroptosis in Breast Cancer Cells through Pyruvate Kinase 2/Caspase-8/Gasdermin C Signaling Pathway. International Journal of Molecular Sciences, 25(19), 10509. https://doi.org/10.3390/ijms251910509