
Effects of Zinc Phthalocyanine Photodynamic Therapy on Vital Structures and Processes in Hela Cells
 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
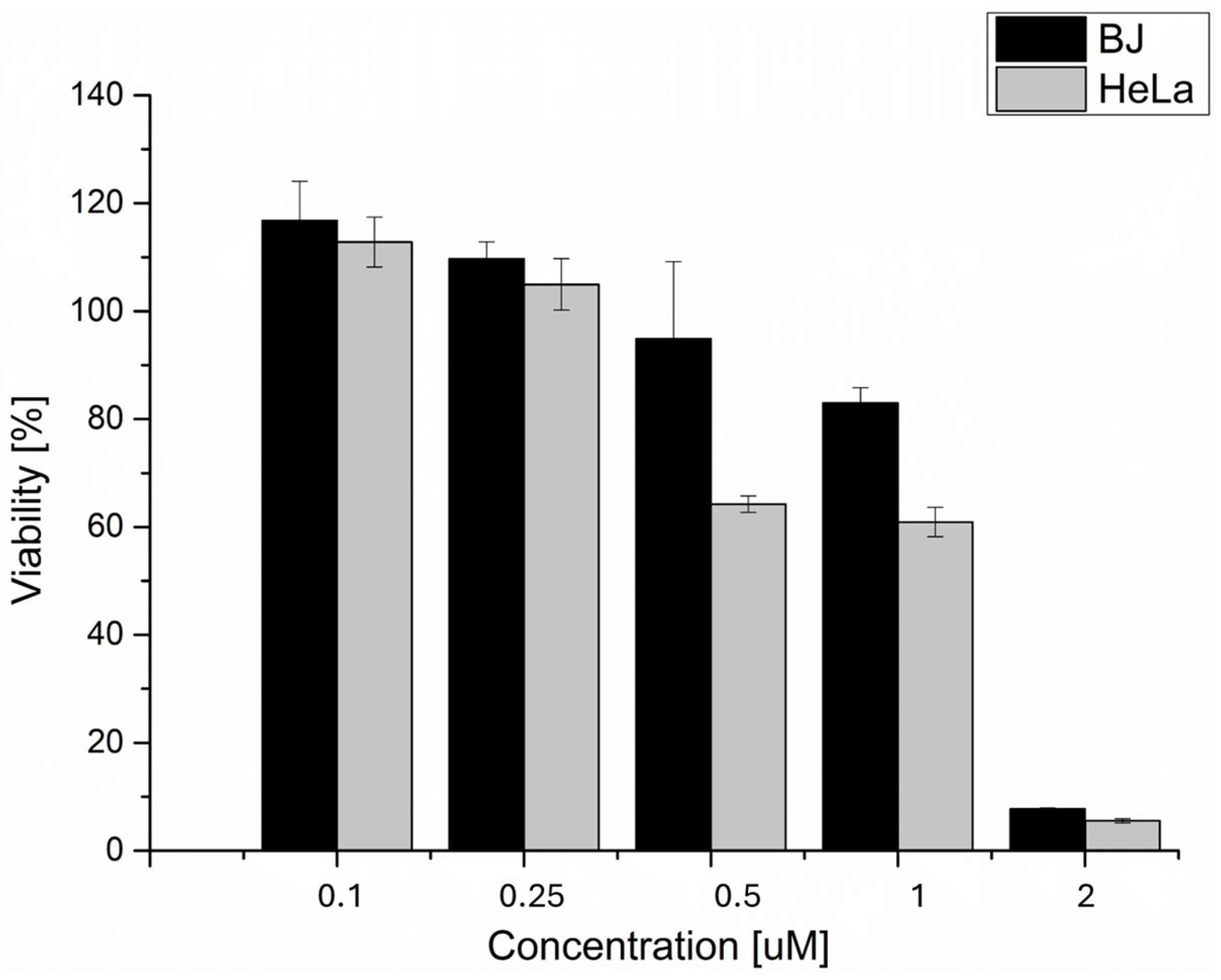
2.1. Cell Viability after 5 Jcm−2 ZnPc PDT Analysis
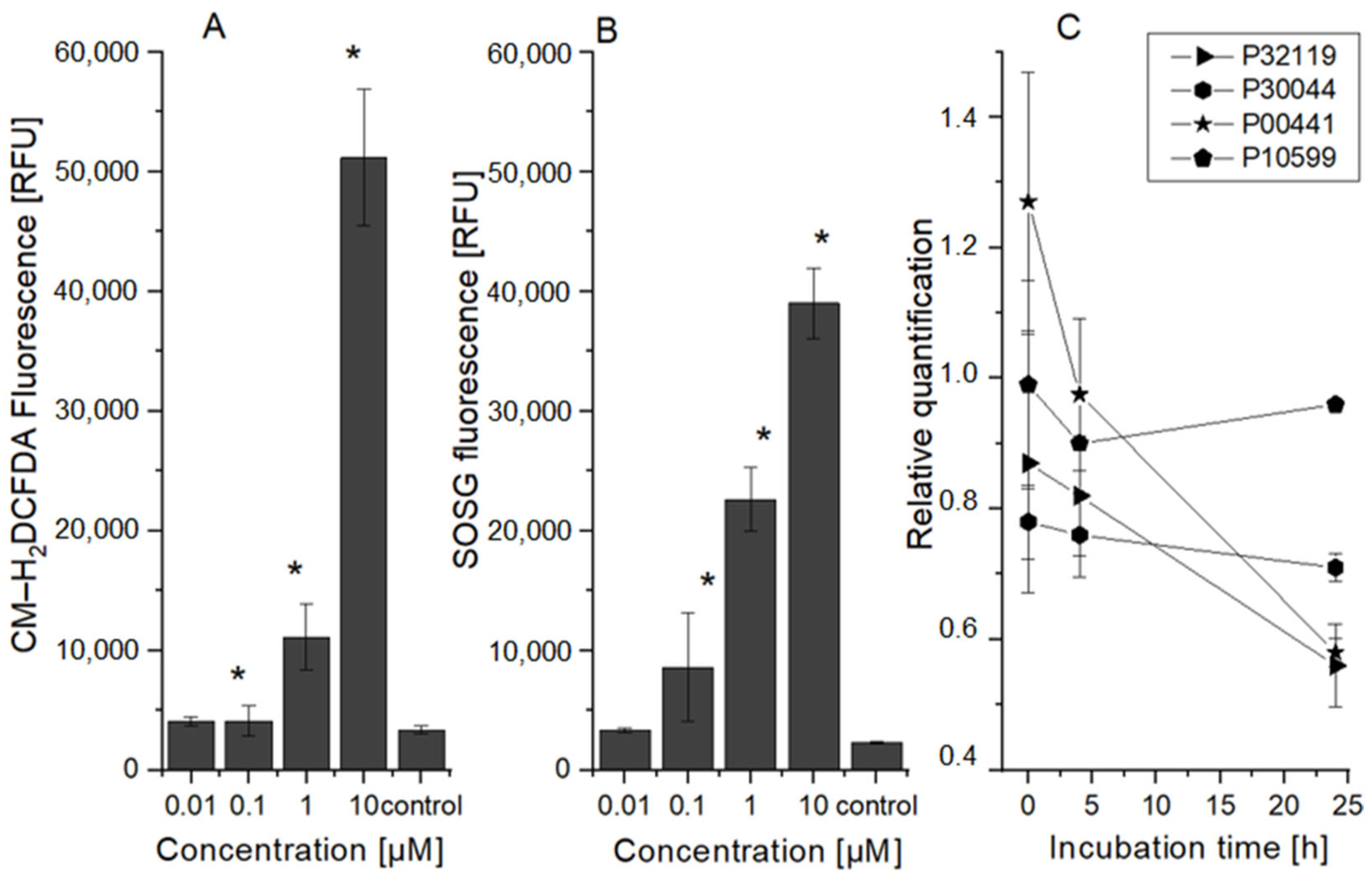
2.2. HeLa Cell Changes at the Oxidative Stress Level
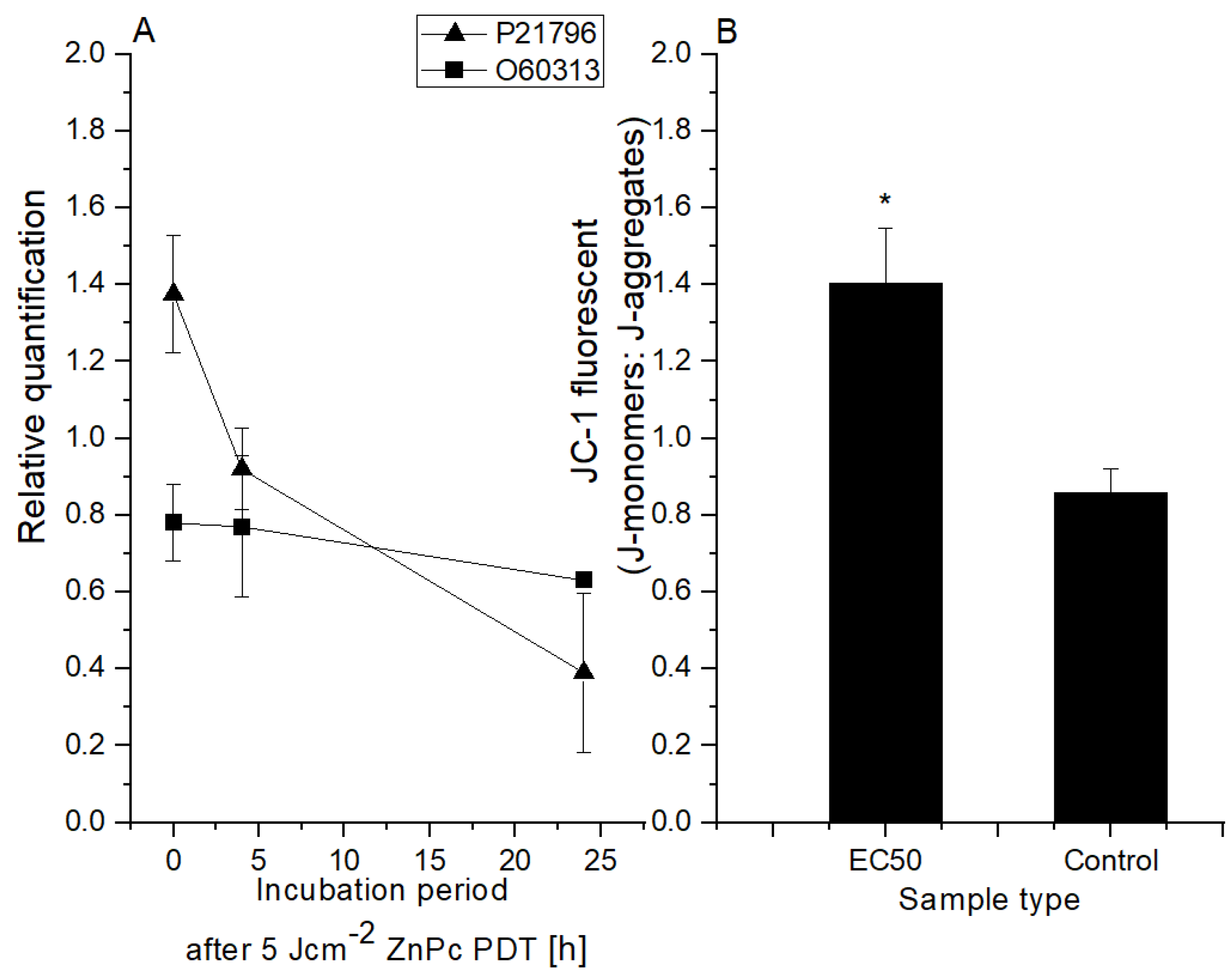
2.3. HeLa Cell Changes at the Mitochondrial Level
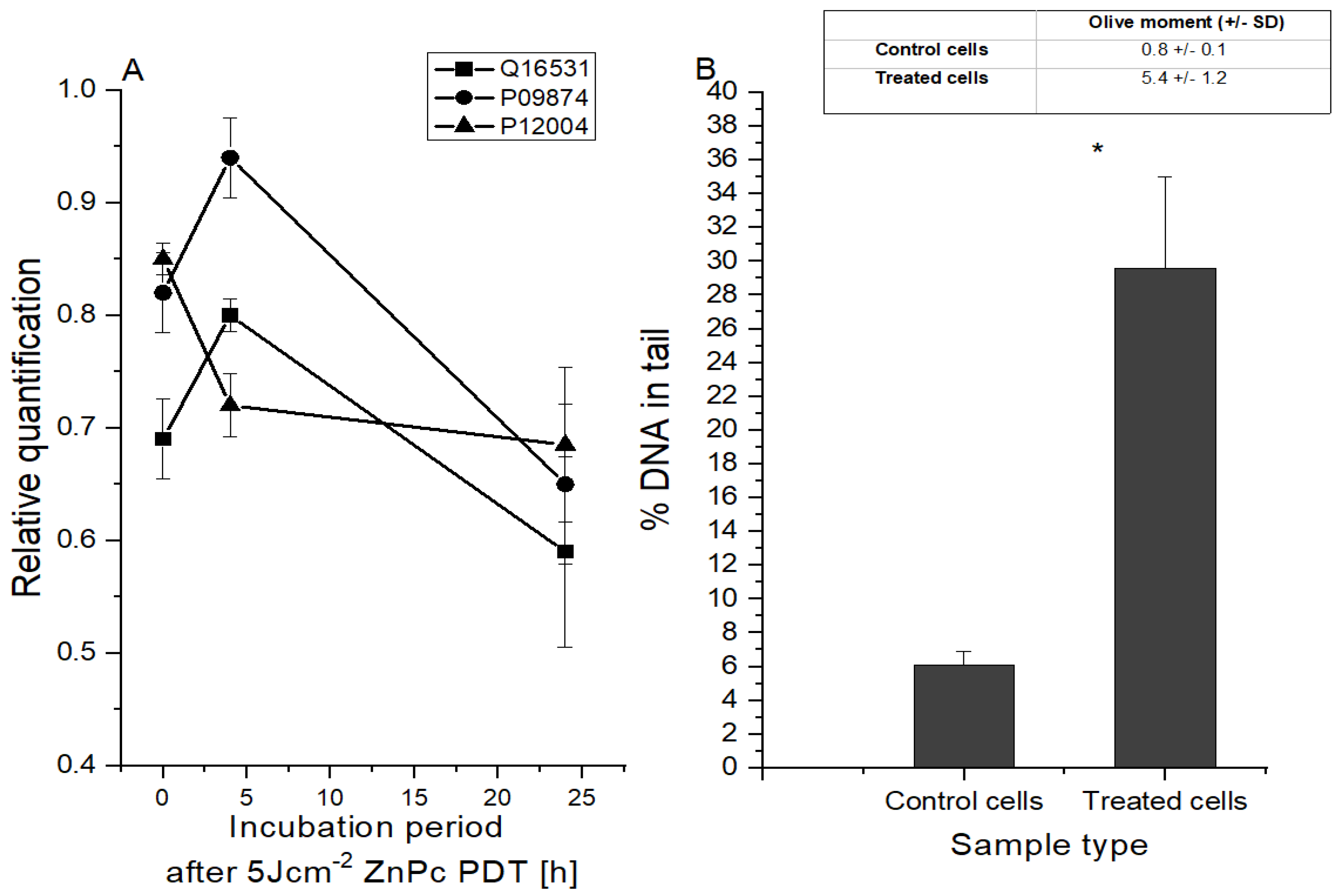
2.4. Changes in HeLa Cells at the DNA Level
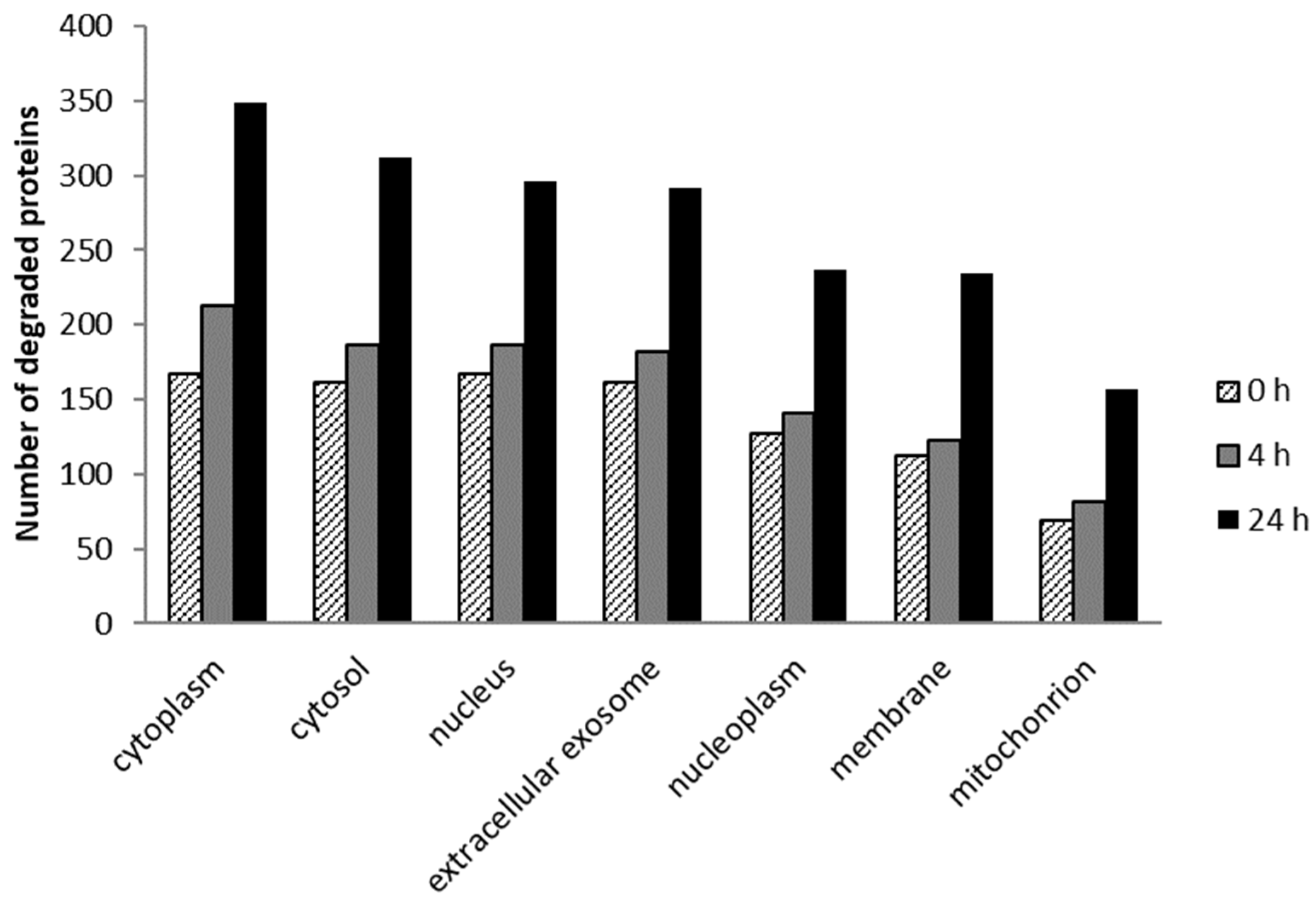
2.5. Cellular Localization and Molecular Function of Degraded Proteins
2.5.1. Cellular Localization
2.5.2. Molecular Function
2.6. Incorporation of the Sensitizer into the Liposomes
3. Materials and Methods
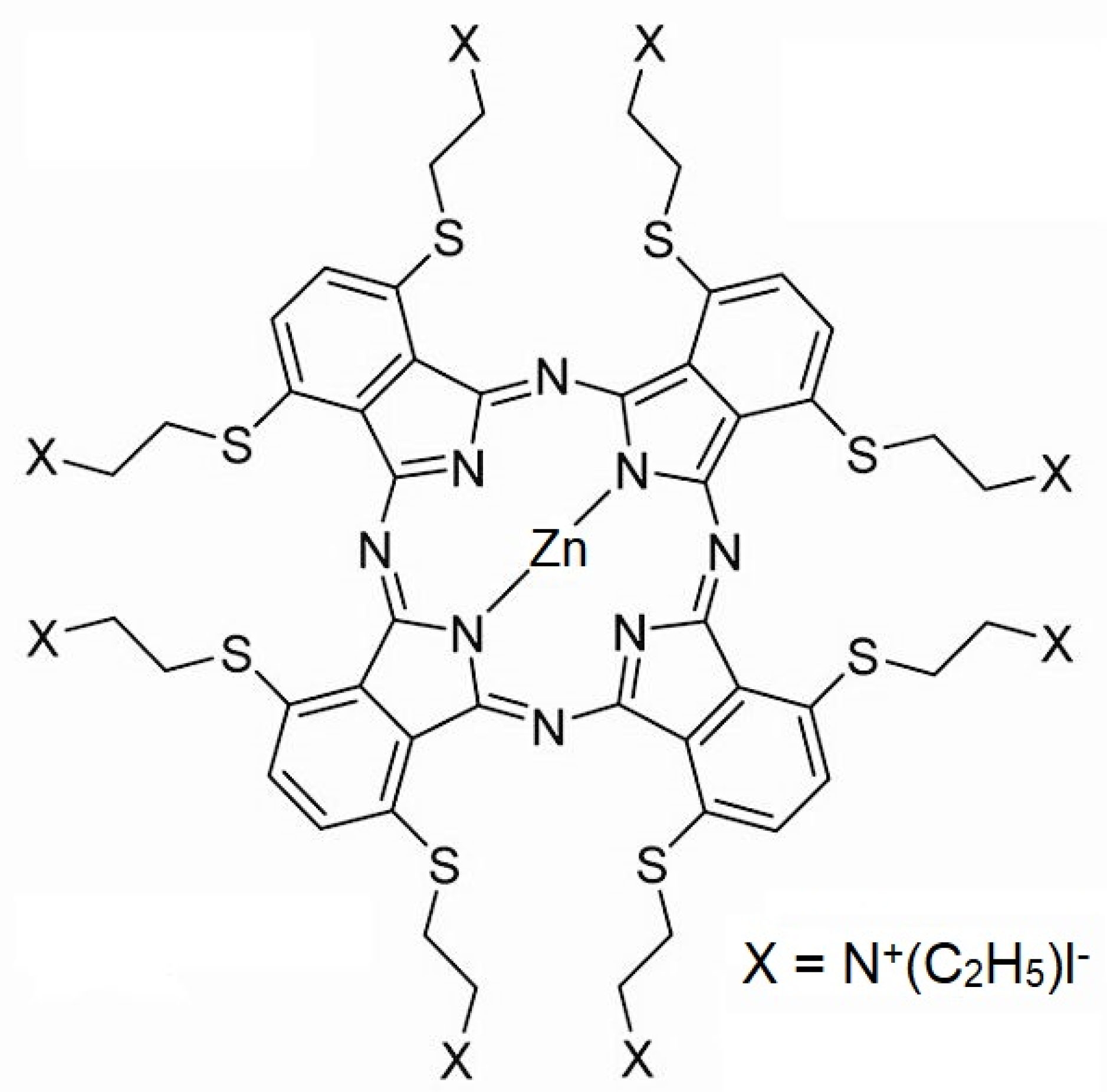
3.1. Photosensitive Substance and Cell Lines
3.2. Light Source and Exposure
3.3. MTT Viability Test
3.4. ROS Measurement
3.4.1. Type I Reaction Products’ Measurement
3.4.2. Type II Reaction Product (Singlet Oxygen) Measurement
3.5. Comet Assay
3.6. Protein Analysis
3.7. Mitochondrial Membrane Potential Measurement
3.8. Incorporation of the Sensitizer into the Liposomes
3.9. Statistical Analysis
4. Conclusions
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Correia, J.H.; Rodrigues, J.A.; Pimenta, S.; Dong, T. Photodynamic Therapy Review: Principles, Photosensitizers, Applications, and Future Directions. Pharmaceutics 2021, 13, 1332. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowaski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kedzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biomechem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Mroz, P.; Bhaumik, J.; Dogutan, D.K.; Aly, Z.; Kamal, Z.; Khalid, L.; Kee, H.L.; Bocian, D.F.; Holten, D.; Lindsey, J.S.; et al. Imidazole metalloporphyrins as photosensitizers for photodynamic therapy: Role of molecular charge, central metal and hydroxyl radical production. Cancer Lett. 2009, 282, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Ochsner, M. Photophysical and photobiological processes in the photodynamic therapy of tumours. J. Photochem. Photobiol. B Biol. 1997, 39, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, N.; Sevilla, A. Current Advances in Photodynamic Therapy (PDT) and the Future Potential of PDT-Combinatorial Cancer Therapies. Int. J. Mol. Sci. 2024, 25, 1023. [Google Scholar] [CrossRef]
- Petrat, F.; Pindiur, S.; Kirsch, M.; de Groot, H. NAD(P)H, a Primary Target of 1O2 in Mitochondria of Intact Cells. J. Biol. Chem. 2003, 278, 3298–3307. [Google Scholar] [CrossRef]
- Petrat, F.; Pindiur, S.; Kirsch, M.; de Groot, H. “Mitochondrial” photochemical drugs do not release toxic amounts of 1O2 within the mitochondrial matrix space. Arch. Biochem. Biophys. 2003, 412, 207–215. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one—Photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef]
- Ma, J.; Jiang, L. Photogeneration of singlet oxygen (1O2) and free radicals (Sen−, O−2) bytetra-brominated hypocrellin B derivative. Free. Radic. Res. 2001, 35, 767–777. [Google Scholar] [CrossRef]
- Nowis, D.; Golab, J. Photodynamic Therapy and Oxidative Stress. In Advances in Photodynamic Therapy—Basic, Translational, and Clinical; Hamblin, M.R., Mróz, P., Eds.; Artech House: New York City, NY, USA, 2008; p. 559. ISBN 978-1-59693-277-7. [Google Scholar]
- Davies, M.J. Singlet oxygen-mediated damage to proteins and its consequences. Biochem. Biophys. Res. Commun. 2003, 305, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Diwanji, N.; Bergmann, A. An unexpected friend—ROS in apoptosis-induced compensatory proliferation: Implications for regeneration and cancer. Semin. Cell Dev. Biol. 2018, 80, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Meng, Z.; Yu, F. The involvement of ROS-regulated programmed cell death in hepatocellular carcinoma. Crit. Rev. Oncol. 2024, 197, 104361. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Tang, Y.; Li, L.; Tao, X. ROS in hepatocellular carcinoma: What we know. Arch. Biochem. Biophys. 2023, 744, 109699. [Google Scholar] [CrossRef]
- Gao, Y.; Huang, D.; Huang, S.; Li, H.; Xia, B. Rational design of ROS generation nanosystems to regulate innate immunity of macrophages, dendrtical and natural killing cells for immunotherapy. Int. Immunopharmacol. 2024, 139, 112695. [Google Scholar] [CrossRef]
- Shah, R.; Ibis, B.; Kashyap, M.; Boussiotis, V.A. The role of ROS in tumor infiltrating immune cells and cancer immunotherapy. Metabolism 2024, 151, 155747. [Google Scholar] [CrossRef]
- Bilski, P.; Motten, A.G.; Bilska, M.; Chignell, C.F. The photooxidation of diethylhydroxylamine by rose bengal in micellar and nonmicellar aqueous solutions. Photochem. Photobiol. 1993, 58, 11–18. [Google Scholar] [CrossRef]
- Xu, D.D.; Leong, M.M.L.; Wong, F.-L.; Lam, H.M.; Hoeven, R.; Yang, Y.; Lv, Q. Photodynamic therapy on prostate cancer cells involve mitochondria membrane proteins. Photodiagnosis Photodyn. Ther. 2020, 31, 101933. [Google Scholar] [CrossRef]
- Xue, L.-Y.; Chiu, S.-M.; Oleinick, N.L. Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene 2001, 20, 3420–3427. [Google Scholar] [CrossRef]
- Jori, G. Far-red-absorbing photosensitizers: Their use in the photodynamic therapy of tumours. J. Photochem. Photobiol. A Chem. 1992, 62, 371–378. [Google Scholar] [CrossRef]
- Wilson, B.C. Photodynamic therapy: Light delivery and dosage for second-generation photosensitizers. Ciba Found Symp. 1989, 146, 60–73; discussion 73–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Li, J.; Yuan, X.; Wang, W.-M.; Xue, J.-P. In vitro photodynamic activities of zinc(II) phthalocyanines substituted with pyridine moieties. Photodiagnosis Photodyn. Ther. 2016, 13, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Hsi, R.A.; Rosenthal, D.I.; Glatstein, E. Photodynamic therapy in the treatment of cancer. Drugs 1999, 54, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Wyss, P.; Schwarz, V.; Dobler-Girdziunaite, D.; Hornung, R.; Walt, H.; Degen, A.; Fehr, M. Photodynamic therapy of locoregional breast cancer recurrences using a chlorin-type photosensitizer. Int. J. Cancer 2001, 93, 720–724. [Google Scholar] [CrossRef]
- Shanazarov, N.A.; Zare, A.; Mussin, N.M.; Albayev, R.K.; Kaliyev, A.A.; Iztleuov, Y.M.; Smailova, S.B.; Tamadon, A. Photodynamic therapy of cervical cancer: A scoping review on the efficacy of various molecules. Ther. Adv. Chronic Dis. 2024, 15, 20406223241233206. [Google Scholar] [CrossRef]
- Fisher, A.B.; Vasquez-Medina, J.P.; Dodia, C.; Sorokina, E.M.; Tao, J.-Q.; Feinstein, S.I. Peroxiredoxin 6 phospholipid hydroperoxidase activity in the repair of peroxidized cell membranes. Redox Biol. 2018, 14, 41–46. [Google Scholar] [CrossRef]
- Li, H.; Benipal, B.; Zhou, S.; Dodia, C.; Chatterjee, S.; Tao, J.-Q.; Sorokina, E.M.; Raabe, T.; Feinstein, S.I.; Fisher, A.B. Critical role of peroxiredoxin 6 in the repair of peroxidized cell membranes following oxidative stress. Free. Radic. Biol. Med. 2015, 87, 356–365. [Google Scholar] [CrossRef]
- Lien, Y.-C.; Feinstein, S.I.; Dodia, C.; Fisher, A.B. The Roles of Peroxidase and Phospholipase A2Activities of Peroxiredoxin 6 in Protecting Pulmonary Microvascular Endothelial Cells Against Peroxidative Stress. Antioxid. Redox Signal. 2012, 16, 440–451. [Google Scholar] [CrossRef]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, Glutaredoxins, and Peroxiredoxins—Molecular Mechanisms and Health Significance: From Cofactors to Antioxidants to Redox Signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Greetham, D.; Vickerstaff, J.; Shenton, D.; Perrone, G.G.; Dawes, I.W.; Grant, C.M. Thioredoxins function as deglutathionylase enzymes in the yeast Saccharomyces cerevisiae. BMC Biochem. 2010, 11, 3. [Google Scholar] [CrossRef]
- Pillay, S.C.; Hofmeyr, J.H.; Rohwer, J.M. The logic of kinetic regulation in the thioredoxin system. BMC Syst. Biol. 2011, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Moroni, M.; Guallini, P.; Torri, C.; Marzatico, F. Antioxidant enzymatic system and free radicals pathway in two different human cancer cell lines. Anticancer Res. 1996, 16, 2741–2747. [Google Scholar] [PubMed]
- Gille, J.J.; Wortelboer, H.M.; Joenje, H. Effect of normobaric hyperoxia on antioxidant defenses of HeLa and CHO cells. Free. Radic. Biol. Med. 1988, 4, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, R.A.; Hernandez, A.; Bloemendal, L.; Ethridge, R.T.; Farrow, B.; Evers, B. Mitochondrial DNA damage and altered membrane potential (ΔΨ) in pancreatic acinar cells induced by reactive oxygen species. Surgery 1999, 126, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Postiglione, I.; Chiaviello, A.; Barra, F.; Roscetto, E.; Soriano, A.A.; Catania, M.R.; Palumbo, G.; Pierantoni, G.M. Mitochondrial Malfunctioning, Proteasome Arrest and Apoptosis in Cancer Cells by Focused Intracellular Generation of Oxygen Radicals. Int. J. Mol. Sci. 2015, 16, 20375–20391. [Google Scholar] [CrossRef]
- Jeong, S.-Y.; Seol, D.-W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef]
- Hsieh, Y.; Wu, C.; Chang, C.; Yu, J. Subcellular localization of Photofrin® determines the death phenotype of human epidermoid carcinoma A431 cells triggered by photodynamic therapy: When plasma membranes are the main targets. J. Cell. Physiol. 2003, 194, 363–375. [Google Scholar] [CrossRef]
- Liu, W.; Vives-Bauza, C.; Acín-Peréz, R.; Yamamoto, A.; Tan, Y.; Li, Y.; Magrané, J.; Stavarache, M.A.; Shaffer, S.; Chang, S.; et al. PINK1 Defect Causes Mitochondrial Dysfunction, Proteasomal Deficit and α-Synuclein Aggregation in Cell Culture Models of Parkinson’s Disease. PLoS ONE 2009, 4, e4597. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Lock, K.; Yenamandra, A.; Cortez, D. DDB1 Maintains Genome Integrity through Regulation of Cdt1. Mol. Cell. Biol. 2006, 26, 7977–7990. [Google Scholar] [CrossRef]
- Hu, J.; McCall, C.M.; Ohta, T.; Xiong, Y. Targeted ubiquitination of CDT1 by the DDB1–CUL4A–ROC1 ligase in response to DNA damage. Nat. Cell Biol. 2004, 6, 1003–1009. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic Therapy. JNCI J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.R.; Holroyd, J.A.; Brown, S.B. The Subcellular Localization of Zn(ll) Phthalocyanines and Their Redistribution on Exposure to Light. Photochem. Photobiol. 1997, 65, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A. Morphologic criteria and detection of apoptosis. Henz 1999, 24, 189–195. [Google Scholar] [CrossRef]
- Alberts, B.; Bray, D.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Základy Buněčné Biologie, 2nd ed.; Espero Publishing, s.r.o.: Prague, Czech Republic, 1998; p. 740. ISBN 80-902906-2-0. [Google Scholar]
- Heider, T.; Kanwan, I.L.; Pandey, V.; Jain, P.; Soni, V. Tumor microenvironment-mediated targeted drug delivery to breast cancer cells. In Targeted Nanomedicine for Breast Cancer Therapy; Elsevier: Amsterdam, The Netherlands, 2022; pp. 305–334. ISBN 978-0-12-824476-0. [Google Scholar]
- Bi, H.; Xue, J.; Jiang, H.; Gao, S.; Yang, D.; Fang, Y.; Shi, K. Current developments in drug delivery with thermosensitive liposomes. Asian J. Pharm. Sci. 2018, 14, 365–379. [Google Scholar] [CrossRef]
- Akram, N.; Afzaal, M.; Saeed, F.; Shah, Y.A.; Faisal, Z.; Asghar, A.; Ateeq, H.; Nayik, G.A.; Wani, S.H.; Hussain, M.; et al. Liposomes: A promising delivery system for active ingredients in food and nutrition. Int. J. Food Prop. 2023, 26, 2476–2492. [Google Scholar] [CrossRef]
- de Matos, M.B.C.; Deckers, R.; van Elburg, B.; Lajoinie, G.; de Miranda, B.S.; Versluis, M.; Schiffelers, R.; Kok, R.J. Ultrasound-Sensitive Liposomes for Triggered Macromolecular Drug Delivery: Formulation and In Vitro Characterization. Front. Pharmacol. 2019, 10, 1463. [Google Scholar] [CrossRef]
- Cidlina, A. Syntéza Kationických Ftalocyaninů. Diploma Thesis, Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Hradec Kralove, Czech Republic, 2011; 72p. [Google Scholar]
- Petřík, I. Analýza Proteinových Fosforylací Proteomickými Metodami. Bachelor’s Thesis, Faculty of Science, Palacky University in Olomouc, Olomouc, Czech Republic, 2013; 62p. [Google Scholar]
- Boersema, P.J.; Raijmakers, R.; Lemeer, S.; Mohammed, S.; Heck, A.J. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 4, 484–494. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | LC50 [µM] ZnPc |
|---|---|
| BJ | 0.09 ± 0.02 |
| HaCat | 0.17 ± 0.03 |
| MCF7 | 0.45 ± 0.17 |
| HeLa | 0.03 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosik, J.; Hosikova, B.; Binder, S.; Lenobel, R.; Kolarikova, M.; Malina, L.; Dilenko, H.; Langova, K.; Bajgar, R.; Kolarova, H. Effects of Zinc Phthalocyanine Photodynamic Therapy on Vital Structures and Processes in Hela Cells. Int. J. Mol. Sci. 2024, 25, 10650. https://doi.org/10.3390/ijms251910650
Hosik J, Hosikova B, Binder S, Lenobel R, Kolarikova M, Malina L, Dilenko H, Langova K, Bajgar R, Kolarova H. Effects of Zinc Phthalocyanine Photodynamic Therapy on Vital Structures and Processes in Hela Cells. International Journal of Molecular Sciences. 2024; 25(19):10650. https://doi.org/10.3390/ijms251910650
Chicago/Turabian StyleHosik, Jakub, Barbora Hosikova, Svatopluk Binder, Rene Lenobel, Marketa Kolarikova, Lukas Malina, Hanna Dilenko, Katerina Langova, Robert Bajgar, and Hana Kolarova. 2024. "Effects of Zinc Phthalocyanine Photodynamic Therapy on Vital Structures and Processes in Hela Cells" International Journal of Molecular Sciences 25, no. 19: 10650. https://doi.org/10.3390/ijms251910650