
Aclarubicin Reduces the Nuclear Mobility of Human DNA Topoisomerase IIβ

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
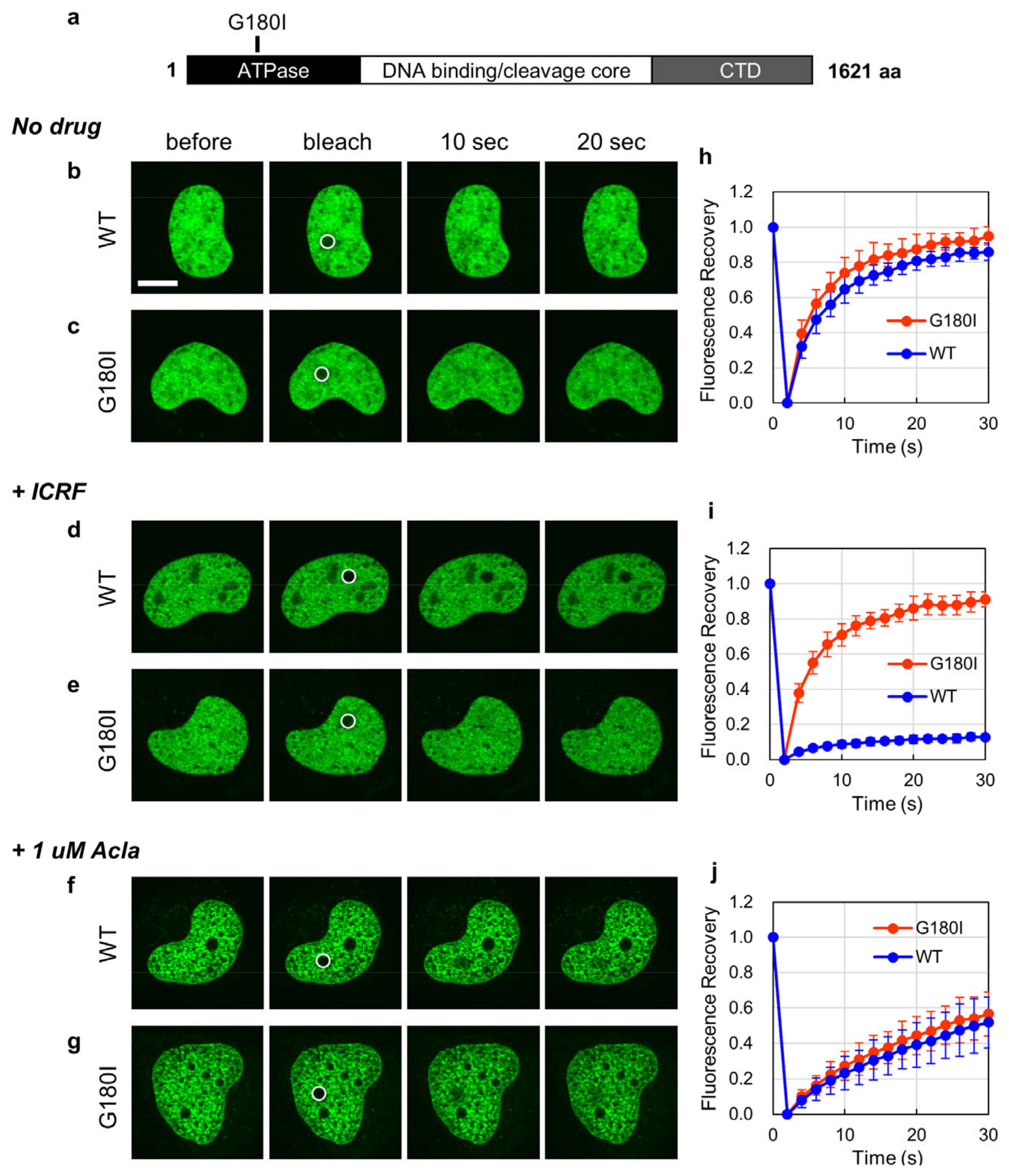
2. Results
2.1. Aclarubicin Inhibits Subnuclear Translocation of TOP2B in ATP-Depleted Cells
2.2. Aclarubicin Reduces the Nuclear Mobility of TOP2B2.1
2.3. Inhibitory Effect of Aclarubicin on TOP2B Mobility Is Independent of TOP2B Enzymatic Activities
2.4. Aclarubicin Reduces Etoposide-Induced DNA Damage
3. Discussion
4. Materials and Methods
4.1. Reagents, Antibodies, and Plasmid
4.2. Cell Culture and Transfection
4.3. ATP Measurement
4.4. Fluorescence Microscopy and Live Cell Imaging
4.5. Immunofluorescence Staining
4.6. FRAP Analysis
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.R.; Ayton, P.; Jones, T.; Davies, S.L.; Simmons, D.L.; Harris, A.L.; Sheer, D.; Hickson, I.D. Isolation of cDNA clones encoding the beta isozyme of human DNA topoisomerase II and localisation of the gene to chromosome 3p24. Nucleic Acids Res. 1992, 20, 5587–5592. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.A.; Sng, J.H.; Patel, S.; Fisher, L.M. Novel HeLa topoisomerase II is the II beta isoform: Complete coding sequence and homology with other type II topoisomerases. Biochim. Biophys. Acta 1993, 1172, 283–291. [Google Scholar] [CrossRef]
- Woessner, R.D.; Mattern, M.R.; Mirabelli, C.K.; Johnson, R.K.; Drake, F.H. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991, 2, 209–214. [Google Scholar]
- Capranico, G.; Tinelli, S.; Austin, C.A.; Fisher, M.L.; Zunino, F. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim. Biophys. Acta 1992, 1132, 43–48. [Google Scholar] [CrossRef]
- Nielsen, C.F.; Zhang, T.; Barisic, M.; Kalitsis, P.; Hudson, D.F. Topoisomerase IIalpha is essential for maintenance of mitotic chromosome structure. Proc. Natl. Acad. Sci. USA 2020, 117, 12131–12142. [Google Scholar] [CrossRef]
- Tiwari, V.K.; Burger, L.; Nikoletopoulou, V.; Deogracias, R.; Thakurela, S.; Wirbelauer, C.; Kaut, J.; Terranova, R.; Hoerner, L.; Mielke, C.; et al. Target genes of Topoisomerase IIbeta regulate neuronal survival and are defined by their chromatin state. Proc. Natl. Acad. Sci. USA 2012, 109, E934–E943. [Google Scholar] [CrossRef]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef]
- Segev, A.; Heady, L.; Crewe, M.; Madabhushi, R. Mapping catalytically engaged TOP2B in neurons reveals the principles of topoisomerase action within the genome. Cell Rep. 2024, 43, 113809. [Google Scholar] [CrossRef]
- Uuskula-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 182. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome Organization Drives Chromosome Fragility. Cell 2017, 170, 507–521.e518. [Google Scholar] [CrossRef]
- Raimer Young, H.M.; Hou, P.C.; Bartosik, A.R.; Atkin, N.D.; Wang, L.; Wang, Z.; Ratan, A.; Zang, C.; Wang, Y.H. DNA fragility at topologically associated domain boundaries is promoted by alternative DNA secondary structure and topoisomerase II activity. Nucleic Acids Res. 2024, 52, 3837–3855. [Google Scholar] [CrossRef]
- Corbett, K.D.; Berger, J.M. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef]
- Drake, F.H.; Zimmerman, J.P.; McCabe, F.L.; Bartus, H.F.; Per, S.R.; Sullivan, D.M.; Ross, W.E.; Mattern, M.R.; Johnson, R.K.; Crooke, S.T.; et al. Purification of topoisomerase II from amsacrine-resistant P388 leukemia cells. Evidence for two forms of the enzyme. J. Biol. Chem. 1987, 262, 16739–16747. [Google Scholar] [CrossRef]
- Drake, F.H.; Hofmann, G.A.; Bartus, H.F.; Mattern, M.R.; Crooke, S.T.; Mirabelli, C.K. Biochemical and pharmacological properties of p170 and p180 forms of topoisomerase II. Biochemistry 1989, 28, 8154–8160. [Google Scholar] [CrossRef]
- Roca, J.; Wang, J.C. DNA transport by a type II DNA topoisomerase: Evidence in favor of a two-gate mechanism. Cell 1994, 77, 609–616. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Vann, K.R.; Oviatt, A.A.; Osheroff, N. Topoisomerase II Poisons: Converting Essential Enzymes into Molecular Scissors. Biochemistry 2021, 60, 1630–1641. [Google Scholar] [CrossRef]
- Roca, J.; Ishida, R.; Berger, J.M.; Andoh, T.; Wang, J.C. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc. Natl. Acad. Sci. USA 1994, 91, 1781–1785. [Google Scholar] [CrossRef]
- Xiao, H.; Mao, Y.; Desai, S.D.; Zhou, N.; Ting, C.Y.; Hwang, J.; Liu, L.F. The topoisomerase IIbeta circular clamp arrests transcription and signals a 26S proteasome pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 3239–3244. [Google Scholar] [CrossRef]
- Murzyn, A.; Orzel, J.; Obajtek, N.; Mroz, A.; Miodowska, D.; Bojdo, P.; Gasiorkiewicz, B.; Koczurkiewicz-Adamczyk, P.; Piska, K.; Pekala, E. Aclarubicin: Contemporary insights into its mechanism of action, toxicity, pharmacokinetics, and clinical standing. Cancer Chemother. Pharmacol. 2024, 94, 123–139. [Google Scholar] [CrossRef]
- Sorensen, B.S.; Sinding, J.; Andersen, A.H.; Alsner, J.; Jensen, P.B.; Westergaard, O. Mode of action of topoisomerase II-targeting agents at a specific DNA sequence. Uncoupling the DNA binding, cleavage and religation events. J. Mol. Biol. 1992, 228, 778–786. [Google Scholar] [CrossRef]
- Sehested, M.; Jensen, P.B. Mapping of DNA topoisomerase II poisons (etoposide, clerocidin) and catalytic inhibitors (aclarubicin, ICRF-187) to four distinct steps in the topoisomerase II catalytic cycle. Biochem. Pharmacol. 1996, 51, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.B.; Sorensen, B.S.; Demant, E.J.; Sehested, M.; Jensen, P.S.; Vindelov, L.; Hansen, H.H. Antagonistic effect of aclarubicin on the cytotoxicity of etoposide and 4′-(9-acridinylamino)methanesulfon-m-anisidide in human small cell lung cancer cell lines and on topoisomerase II-mediated DNA cleavage. Cancer Res. 1990, 50, 3311–3316. [Google Scholar]
- Holm, B.; Jensen, P.B.; Sehested, M.; Hansen, H.H. In vivo inhibition of etoposide-mediated apoptosis, toxicity, and antitumor effect by the topoisomerase II-uncoupling anthracycline aclarubicin. Cancer Chemother. Pharmacol. 1994, 34, 503–508. [Google Scholar] [CrossRef]
- Gajek, A.; Rogalska, A.; Koceva-Chyla, A. Aclarubicin in subtoxic doses reduces doxorubicin cytotoxicity in human non-small cell lung adenocarcinoma (A549) and human hepatocellular carcinoma (HepG2) cells by decreasing DNA damage. Toxicol. In Vitro 2019, 55, 140–150. [Google Scholar] [CrossRef]
- Richard, D.; Hollender, P.; Chenais, B. Involvement of reactive oxygen species in aclarubicin-induced differentiation and invasiveness of HL-60 leukemia cells. Int. J. Oncol. 2002, 21, 393–399. [Google Scholar] [CrossRef]
- Rogalska, A.; Koceva-Chyla, A.; Jozwiak, Z. Aclarubicin-induced ROS generation and collapse of mitochondrial membrane potential in human cancer cell lines. Chem. Biol. Interact. 2008, 176, 58–70. [Google Scholar] [CrossRef]
- Wooten, M.; Takushi, B.; Ahmad, K.; Henikoff, S. Aclarubicin stimulates RNA polymerase II elongation at closely spaced divergent promoters. Sci. Adv. 2023, 9, eadg3257. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.A.; Kanellis, D.C.; Saproo, S.; Leal, K.; Martinez, J.F.; Bartek, J.; Lindstrom, M.S. Chromatin damage generated by DNA intercalators leads to degradation of RNA Polymerase II. Nucleic Acids Res. 2024, 52, 4151–4166. [Google Scholar] [CrossRef] [PubMed]
- Nesher, E.; Safina, A.; Aljahdali, I.; Portwood, S.; Wang, E.S.; Koman, I.; Wang, J.; Gurova, K.V. Role of Chromatin Damage and Chromatin Trapping of FACT in Mediating the Anticancer Cytotoxicity of DNA-Binding Small-Molecule Drugs. Cancer Res. 2018, 78, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4, 1908. [Google Scholar] [CrossRef]
- Pang, B.; de Jong, J.; Qiao, X.; Wessels, L.F.; Neefjes, J. Chemical profiling of the genome with anti-cancer drugs defines target specificities. Nat. Chem. Biol. 2015, 11, 472–480. [Google Scholar] [CrossRef]
- Christensen, M.O.; Larsen, M.K.; Barthelmes, H.U.; Hock, R.; Andersen, C.L.; Kjeldsen, E.; Knudsen, B.R.; Westergaard, O.; Boege, F.; Mielke, C. Dynamics of human DNA topoisomerases IIalpha and IIbeta in living cells. J. Cell Biol. 2002, 157, 31–44. [Google Scholar] [CrossRef]
- Onoda, A.; Hosoya, O.; Sano, K.; Kiyama, K.; Kimura, H.; Kawano, S.; Furuta, R.; Miyaji, M.; Tsutsui, K.; Tsutsui, K.M. Nuclear dynamics of topoisomerase IIbeta reflects its catalytic activity that is regulated by binding of RNA to the C-terminal domain. Nucleic Acids Res. 2014, 42, 9005–9020. [Google Scholar] [CrossRef]
- Morotomi-Yano, K.; Yano, K.I. Nucleolar translocation of human DNA topoisomerase II by ATP depletion and its disruption by the RNA polymerase I inhibitor BMH-21. Sci. Rep. 2021, 11, 21533. [Google Scholar] [CrossRef]
- Morotomi-Yano, K.; Hiromoto, Y.; Higaki, T.; Yano, K.I. Disease-associated H58Y mutation affects the nuclear dynamics of human DNA topoisomerase IIbeta. Sci. Rep. 2022, 12, 20627. [Google Scholar] [CrossRef]
- Skouboe, C.; Bjergbaek, L.; Oestergaard, V.H.; Larsen, M.K.; Knudsen, B.R.; Andersen, A.H. A human topoisomerase II alpha heterodimer with only one ATP binding site can go through successive catalytic cycles. J. Biol. Chem. 2003, 278, 5768–5774. [Google Scholar] [CrossRef]
- Jensen, S.; Andersen, A.H.; Kjeldsen, E.; Biersack, H.; Olsen, E.H.; Andersen, T.B.; Westergaard, O.; Jakobsen, B.K. Analysis of functional domain organization in DNA topoisomerase II from humans and Saccharomyces cerevisiae. Mol. Cell Biol. 1996, 16, 3866–3877. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Lobrich, M.; Shiloh, Y.; Chen, D.J. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; van der Zanden, S.Y.; Wander, D.P.A.; Borras, D.M.; Song, J.Y.; Li, X.; van Duikeren, S.; van Gils, N.; Rutten, A.; van Herwaarden, T.; et al. Uncoupling DNA damage from chromatin damage to detoxify doxorubicin. Proc. Natl. Acad. Sci. USA 2020, 117, 15182–15192. [Google Scholar] [CrossRef] [PubMed]
- Manville, C.M.; Smith, K.; Sondka, Z.; Rance, H.; Cockell, S.; Cowell, I.G.; Lee, K.C.; Morris, N.J.; Padget, K.; Jackson, G.H.; et al. Genome-wide ChIP-seq analysis of human TOP2B occupancy in MCF7 breast cancer epithelial cells. Biol. Open 2015, 4, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- van Ruiten, M.S.; Rowland, B.D. On the choreography of genome folding: A grand pas de deux of cohesin and CTCF. Curr. Opin. Cell Biol. 2021, 70, 84–90. [Google Scholar] [CrossRef]
- de Wit, E.; Nora, E.P. New insights into genome folding by loop extrusion from inducible degron technologies. Nat. Rev. Genet. 2023, 24, 73–85. [Google Scholar] [CrossRef]
- Gittens, W.H.; Johnson, D.J.; Allison, R.M.; Cooper, T.J.; Thomas, H.; Neale, M.J. A nucleotide resolution map of Top2-linked DNA breaks in the yeast and human genome. Nat. Commun. 2019, 10, 4846. [Google Scholar] [CrossRef]
- Hansen, A.S.; Pustova, I.; Cattoglio, C.; Tjian, R.; Darzacq, X. CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife 2017, 6, e25776. [Google Scholar] [CrossRef]
- Mazza, D.; Abernathy, A.; Golob, N.; Morisaki, T.; McNally, J.G. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. 2012, 40, e119. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Z.; Li, L.; Chen, B.C.; Revyakin, A.; Hajj, B.; Legant, W.; Dahan, M.; Lionnet, T.; Betzig, E.; et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 2014, 156, 1274–1285. [Google Scholar] [CrossRef]
- Liu, Z.; Tjian, R. Visualizing transcription factor dynamics in living cells. J. Cell Biol. 2018, 217, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. Protein dynamics: Implications for nuclear architecture and gene expression. Science 2001, 291, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Morotomi-Yano, K.; Saito, S.; Adachi, N.; Yano, K.I. Dynamic behavior of DNA topoisomerase IIbeta in response to DNA double-strand breaks. Sci. Rep. 2018, 8, 10344. [Google Scholar] [CrossRef]
- Britton, S.; Coates, J.; Jackson, S.P. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2013, 202, 579–595. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Khan, T.; Kandola, T.S.; Wu, J.; Venkatesan, S.; Ketter, E.; Lange, J.J.; Rodriguez Gama, A.; Box, A.; Unruh, J.R.; Cook, M.; et al. Quantifying Nucleation In Vivo Reveals the Physical Basis of Prion-like Phase Behavior. Mol. Cell 2018, 71, 155–168.e157. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morotomi-Yano, K.; Yano, K.-i. Aclarubicin Reduces the Nuclear Mobility of Human DNA Topoisomerase IIβ. Int. J. Mol. Sci. 2024, 25, 10681. https://doi.org/10.3390/ijms251910681
Morotomi-Yano K, Yano K-i. Aclarubicin Reduces the Nuclear Mobility of Human DNA Topoisomerase IIβ. International Journal of Molecular Sciences. 2024; 25(19):10681. https://doi.org/10.3390/ijms251910681
Chicago/Turabian StyleMorotomi-Yano, Keiko, and Ken-ichi Yano. 2024. "Aclarubicin Reduces the Nuclear Mobility of Human DNA Topoisomerase IIβ" International Journal of Molecular Sciences 25, no. 19: 10681. https://doi.org/10.3390/ijms251910681